Workflow management
Week 9 – Lecture A

1 Introduction
1.1 Learning goals
This week
This week, we’ll cover two distinct topics:
- Workflow management (this session)
- Data management and sharing (next session)
This session
- What a Markdown protocol of your workflow can look like
- How you can automate such workflows with Bash and Slurm, and what the associated challenges are
- What “workflow management systems” are, and what the advantages of formal pipelines/workflows written with these are
- That you may be able to use publicly available pipelines such as those produced by the nf-core initiative
1.2 Getting ready
- At https://ondemand.osc.edu, start a VS Code session in
/fs/ess/PAS2880/users/$USER
2 Structuring your analysis workflow
2.1 Multi-step workflows
In a typical research project with omics data (and likely in your final project for this course), you need to run multiple consecutive steps to process/analyze your data. In other words, you will have a “workflow” that you need to structure properly and reproducibly.
In the past few weeks, we have focused on writing code to complete individual steps of such workflows. Now, we’ll zoom out to look at the bigger picture: how do you organize and run your workflows as a whole?
Once again, let’s look at this RNA-Seq analysis workflow you’ve seen before, going from raw reads to gene counts:
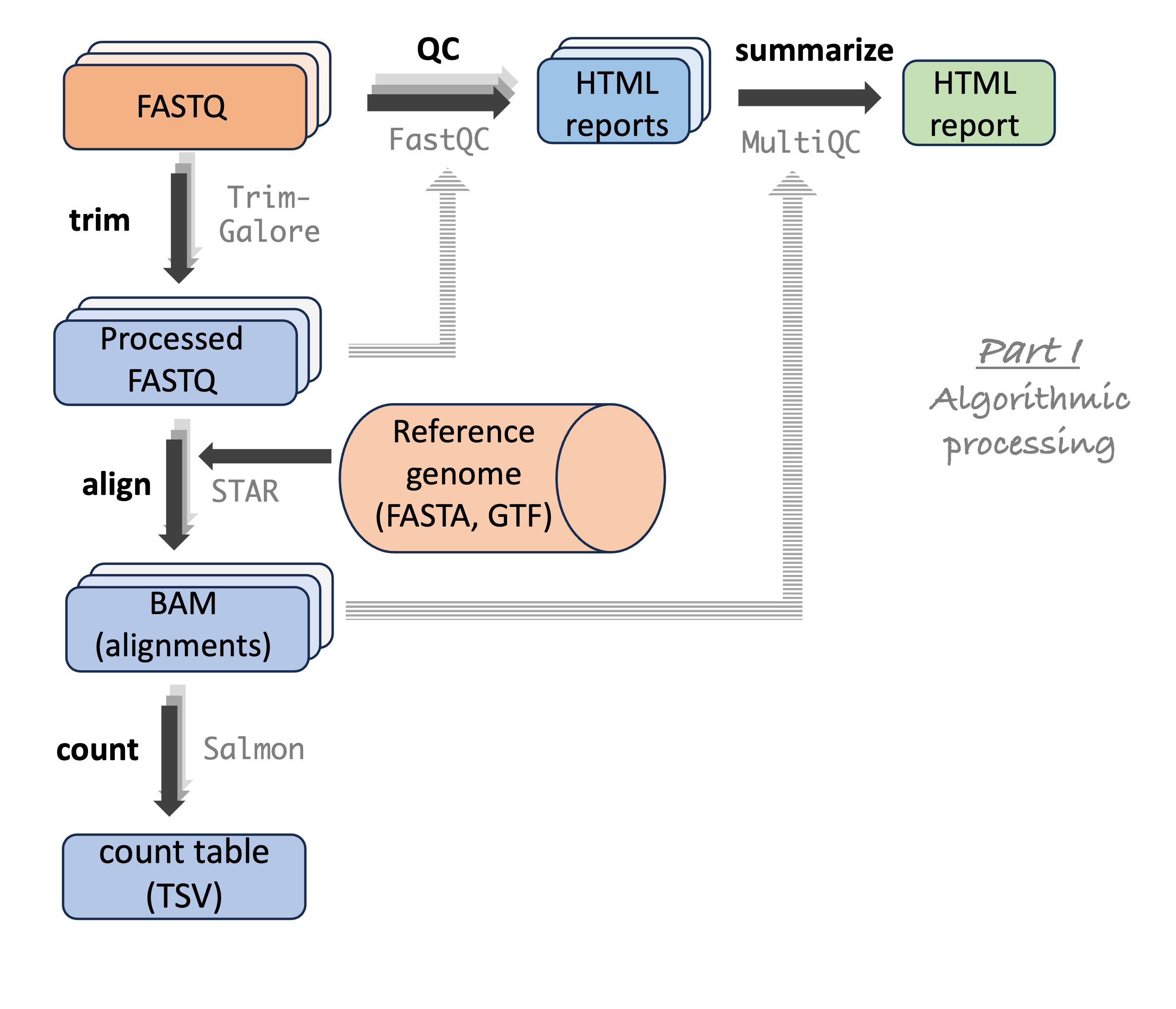
In the past few weeks of this course, you’ve created Markdown files with the commands you used to run your scripts. For example, these files contained for loops to submit many FastQC and TrimGalore batch jobs.
As discussed in week 2, for larger projects, it is a good idea to create at least two separate Markdown files for the code you used to run scripts and other commands:
A “lab notebook”-style file that has chronological entries and is comprehensive: this should cover everything of note that you did, including mishaps, dead ends, and things that were redone differently later on.
A “protocol” (or “recipe” / “workflow”)-style file that has the final code in order of execution needed to run and rerun your entire project’s computational work.
Let’s see a more worked-out example of a protocol file.
2.2 A longer protocol example
Specifically, we’ll include the following steps:
- Read QC with FastQC (independently for each file)
- FastQC summary with MultiQC (once for all files)
- Read trimming with TrimGalore (independently for each sample)
- Trimmed read alignment to a reference genome with STAR (independently for each sample)
- Gene-level alignment counting with Salmon (independently for each sample)
# RNA-Seq analysis of _Culex pipiens_
- Author: Jelmer Poelstra, MCIC, OSU (poelstra.1@osu.edu)
- Date: First created on 2025-04-01, last modified on 2025-08-25
- GitHub repository: <https://github.com/jelmerp/culex>
- This was run at the Pitzer cluster of the Ohio Supercomputer Center (<www.osc.edu>)
- OSC working dir: /fs/ess/PAS2880/users/jelmer
- More information about the project and its files:
see the `README.md` file in the abovementioned working dir
## Prerequisites
- **Input files**:
- The input files are:
- Paired-end FASTQ files
- Reference genomes files `assembly.fna` and `annotation.gtf`
- Input file paths are set below and can be adjusted to your situation.
- If you're running this at OSC and have access to project PAS2880,
you can copy the files from the working dir specified above,
or adjust the input file paths below to directly use these files.
- Otherwise:
- Reference genome files can be downloaded from <URL>
- FASTQ files are available through the SRA via accession number <ACCESSION>
- **Software**:
- The scripts will run programs using Singularity/Apptainer containers
available online, with links contained inside the scripts
- If running at OSC, no software installations are needed
- If you're not at OSC, you need to have `apptainer` and `Slurm` installed.
- You don't need to create the output dirs because the scripts will do so.<!--- [Hypothetical example - don't run any of the code in this document] -->
## Set up: Set the input file paths
```bash
fastq_dir=data/fastq
ref_assembly=results/01_refseqs/assembly.fna
ref_annotation=results/01_refseqs/annotation.gtf
```
## Step 1: Read QC with FastQC
The `fastqc.sh` script runs on 1 FASTQ file at a time and takes 2 arguments -
the input FASTQ file and output dir:
```bash
for fastq in "$fastq_dir"/*fastq.gz; do
sbatch scripts/fastqc.sh "$fastq" results/fastqc
done
```
## Step 2: FastQC summary with MultiQC
The `multiqc.sh` script runs on all FastQC outputs at a time and takes 2 arguments -
the input dir (i.e., the FastQC output dir) and output dir:
```bash
sbatch scripts/multiqc.sh results/fastqc results/multiqc
```
## Step 3: Read trimming with TrimGalore
The `trim.sh` script runs for one sample at a time and takes 3 arguments -
input forward-read (R1) FASTQ, reverse-read (R2) FASTQ, and the desired output dir:
```bash
for R1 in "$fastq_dir"/*_R1.fastq.gz; do
R2=${R1/_R1/_R2}
sbatch scripts/trimgalore.sh "$R1" "$R2" results/trimgalore
done
```
## Step 3: Trimmed read alignment to a reference genome with STAR
The `star.sh` script runs for one sample at a time and takes 4 arguments -
input trimmed R1 and R2 FASTQs produced by `trimgalore.sh`, a reference FASTA file,
and the desired output dir:
```bash
for R1 in results/trimgalore/*_R1.fastq.gz; do
R2=${R1/_R1/_R2}
sbatch scripts/star.sh "$R1" "$R2" "$ref_assembly" results/star
done
```
## Step 5: Gene-level alignment counting with Salmon
The `salmon.sh` script runs for one sample at a time and takes 4 arguments -
input BAM file produced by `start.sh`, a reference GTF file,
and the desired output dir:
```bash
for bam in results/star/*bam; do
sbatch scripts/salmon.sh "$bam" "$ref_annotation" results/salmon
done
```2.3 Why organize your workflow this way?
The protocol above submits a shell script one or more times for each step, with each script taking arguments to specify the in- and output files, and running a bioinformatics program to perform that step.
Here are some advantages of structuring your workflows with single-step, flexible (i.e., argument-accepting) scripts, and an overarching protocol Markdown file to describe the entire workflow:
- Rerunning everything, including with a modified sample set, or tool settings, is relatively straightforward – both for yourself and others, improving reproducibility
- Re-applying the same set of analyses in a different project is straightforward
- The Markdown file documents the steps taken
- It (more or less) ensures you are including all necessary steps
However, in the example above, it is import to realize that you cannot run all the code at once.
Why can’t all the code be run at once? (Click to see the solution)
This is because most steps need the input with both the second and third steps using the output of an earlier step as their input.
Therefore, if you run all code at once, all batch jobs will be submitted at the same time: this means that later steps will start running before the earlier step has finished, i.e. before their input files are available, and will fail.Which steps can be run simultaneously? (Click to see the solution)
This is because most steps need the input with both the second and third steps using the output of an earlier step as their input.2.4 How about automation?
The above protocol is not an automated pipeline because for most steps, you can’t run them until the previous step has finished. You may therefore consider trying to improve automation, which you can do with two main approaches:
- With more advanced Bash and Slurm code
- With “workflow management systems” like Nextflow (recommended but takes some learning)
In this course, we will cover neither approach in detail1, but the rest of this lecture serves to at least make you aware of the possibilities. This is a more advanced topic that I would recommend to those of you planning to work with omics (especially genomics and transcriptomics) data a lot.
3 Improving automation with Bash and Slurm
When trying to improve automation with Bash and Slurm, here are two approaches to potentially consider:
- Reducing the number of steps by creating multi-step shell scripts and/or
- Automatically managing batch job submissions with job “dependencies”
We’ll go over each of these below.
3.1 Reduce the number of steps by combining scripts
At the extreme, you could write a single shell script for all steps and all samples (i.e., a script with multiple loops) that can be submitted as a single batch job. While this would work, it would be highly inefficient2 because sample-wise steps would run consecutively across samples instead of simultaneously.
A more reasonable approach would be to write scripts that only process one sample but include as many steps as possible for that sample.
However, this has downsides too, some of which can be overcome with a further increase in script complexity. For example, what if you need to redo only the steps from the middle of your workflow onwards (e.g. because you want to change some settings there). Rerunning your all-inclusive script would mean rerunning all steps, which could be a big waste of time. You could overcome this by including clauses in your script to only rerun certain steps – see the Appendix.
At that point, though, the increased amount of time spent on writing your scripts and the general burden of having to maintain more complicated scripts may not be worth the time you save with increased automation. All in all, I personally prefer to write shell scripts that just run one program and for one sample, where appropriate.
3.2 Automatically manage batch job submission
There are ways to automate Slurm batch job submission so you don’t have to wait for each step to finish before moving on to the next. Specifically, you can specify so-called Slurm “job dependencies” that only allow a job to to actually start once one or more other jobs have (sucessfully) finished running. For an example, see the Appendix.
However, similar to the approach with longer shell scripts, the added complexity comes with its own costs, and may only be able to improve automation somewhat. Therefore, it may in the end not pay off.
4 Automation with workflow management systems
4.1 Introduction
Pipeline/workflow tools, often called “workflow management systems” in full, provide ways to formally describe and execute pipelines.

A quote from this article, Perkel (2019):
Workflows can involve hundreds to thousands of data files; a pipeline must be able to monitor their progress and exit gracefully if any step fails. And pipelines must be smart enough to work out which tasks need to be re-executed and which do not.
The two most commonly used command-line based options in bioinformatics are Nextflow (Di Tommaso et al. (2017)) and Snakemake (Mölder et al. (2025)). Both are small “domain-specific” languages (DSLs) that are an extension of a more general language: Python for Snakemake, and Groovy/Java for Nextflow.
4.2 Pros and cons of workflow management systems
The advantages of using workflow management systems rather than some of the abovementioned approaches are improved automation, flexibility (e.g., ease of running with different settings or on different datasets), portability (ease of running on different computers), and scalability (ease of scaling up to larger datasets):
- Automation
- Detect & rerun upon changes in input files and failed steps.
- Automate Slurm job submissions.
- Integration with software management.
- Flexibility, portability, and scalability
This is due to these tools separating generic pipeline nuts-and-bolts from the following two aspects:- Run-specific configuration — samples, directories, settings/parameters.
- Things specific to the run-time environment (laptop vs. cluster vs. cloud).
The main disadvantage is that they have a somewhat steep learning curve and pipelines are more complicated to write than you might expect for a tool that “merely” ties your workflow steps together.
But overall, my recommendation with respect to workflow management is to either:
- Accept that you’re workflow isn’t automated, and use Markdown protocols like the one above
- Go all-in and use a workflow tool (I recommend Nextflow, specifically)
5 Pre-existing pipelines
Another thing to keep in mind is that you don’t always have to build your own workflow/pipeline. Many publicly available pipelines exist. And if reliable pipelines exist for your planned analyses, there are several advantages to using one of these instead of trying to build your own pipeline.
For example, a bewildering array of bioinformatics programs is often available for a given type of analysis, and it can be very hard and time-consuming to figure out what you should use. With pipelines from certain initiatives, you can be confident that it uses a good combination of tools and tool settings: most of these pipelines have been developed over years by many experts in the field, and are continuously updated.
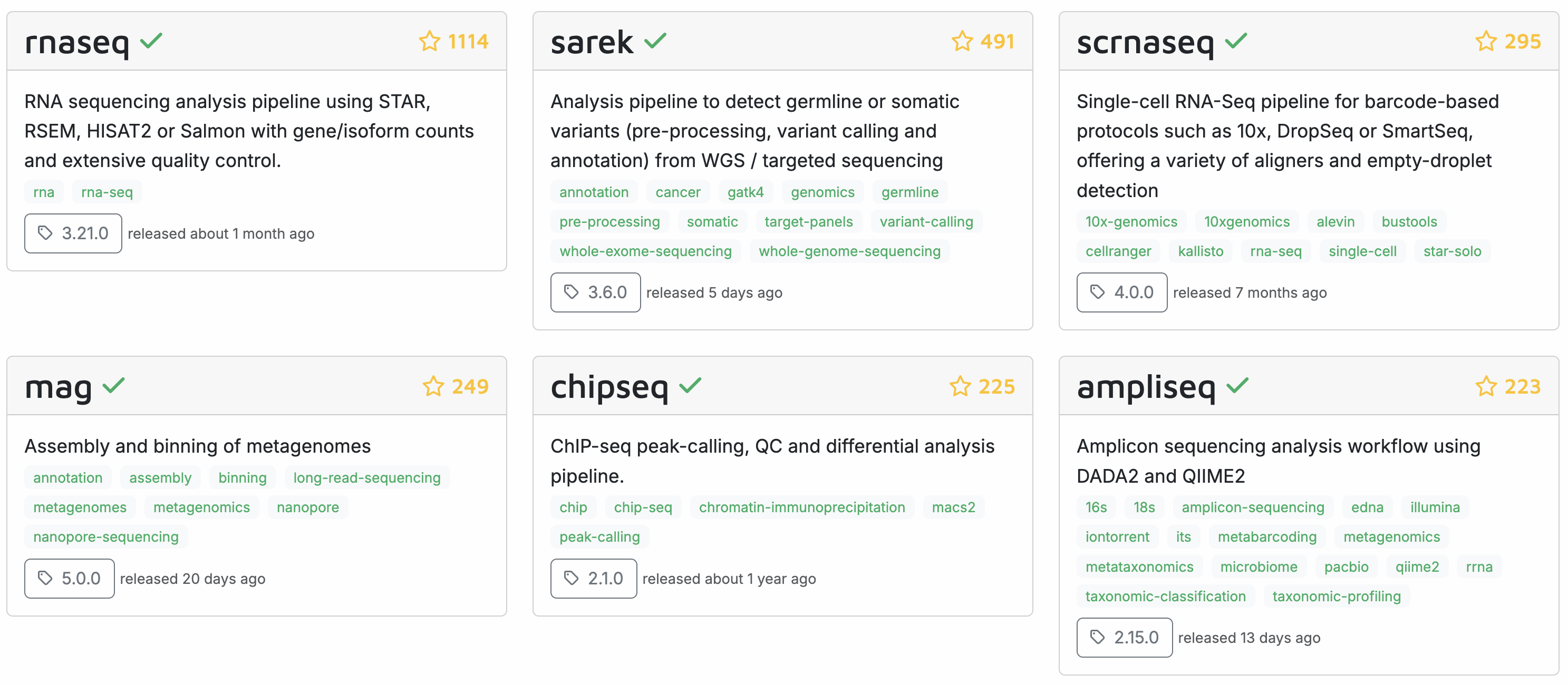
Among workflow tools, Nextflow has by far the best ecosystem of publicly available pipelines. The “nf-core” initiative (https://nf-co.re, Ewels et al. (2020)) curates a set of best-practice, highly flexible, and well-documented pipelines written in Nextflow:

For many common omics analysis types, nf-core has a pipeline. It currently has 84 complete pipelines – these are the six most popular ones:
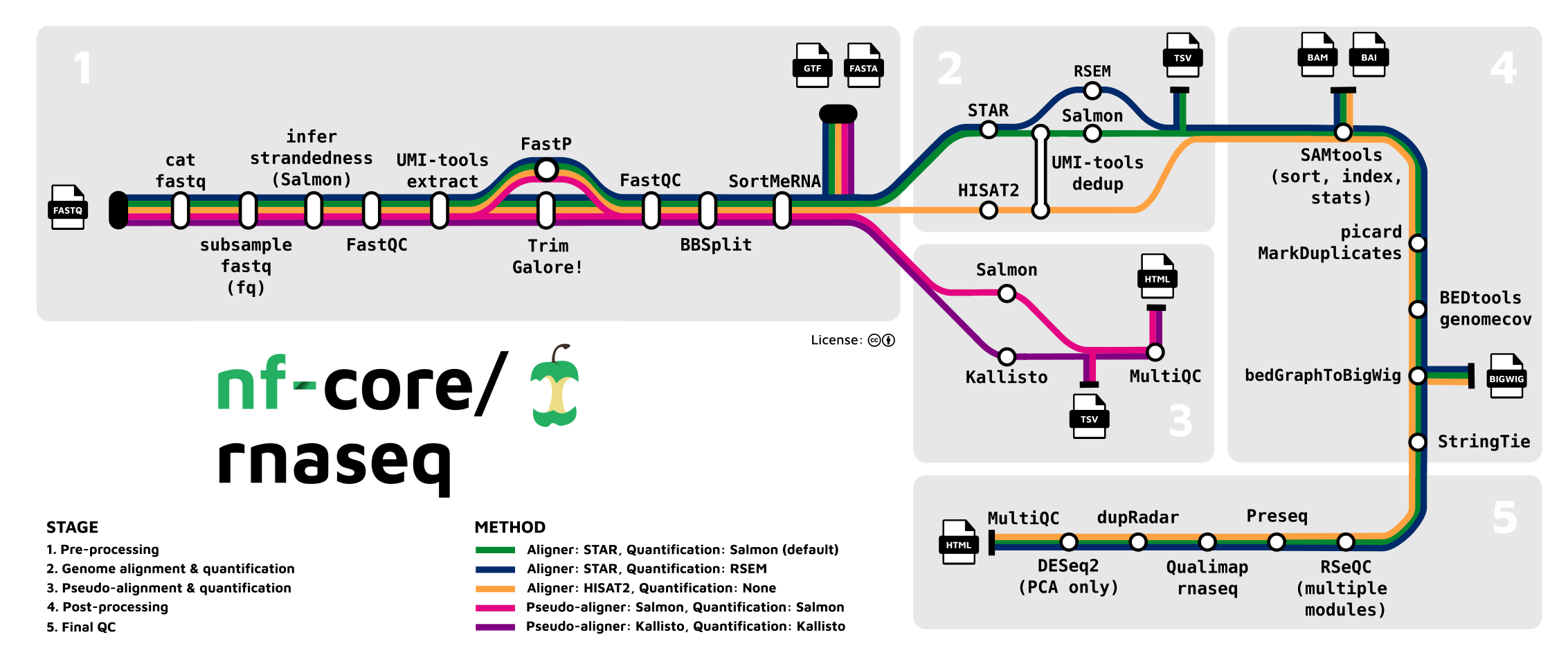
Here’s a closer look at the most widely used nf-core pipeline, the rnaseq pipeline, which we’ll run in week 15 of the course:
6 Appendix: automation with Bash and Slurm
6.1 if-statements
if-statements will allow you to flexibly run part of a script, for example:
trim=$1 # true or false
map=$2 # true or false
count=$3 # true or false
if [[ "$trim" == true ]]; then
for R1 in data/fastq/*_R1.fastq.gz; do
bash scripts/trim.sh "$R1" results/trim
done
fi
if [[ "$map" == true ]]; then
for R1 in results/trim/*_R1.fastq.gz; do
bash scripts/map.sh "$R1" results/map
done
fi
if [[ "$count" == true ]]; then
bash scripts/count.sh results/map results/count_table.txt
fiTo learn more about if statements, see this section of the bonus page on shell scripting.
6.2 Script options
If your script has a whole bunch of options to specify which steps should be run, it will become tedious and error prone to work with the regular “positional arguments”. Instead, you should then consider writing your script such that it can accept options too, e.g.:
bash myscripts.sh --skip_count --skip_map $fastq $outdirTo learn how to do this, see this section of the bonus page on shell scripting.
6.3 Slurm job dependencies
In the example below, jobs will only start after their “dependencies” (jobs whose outputs they need) have finished:
for R1 in data/fastq/*_R1.fastq.gz; do
# Submit the trimming job and store its job number:
submit_line=$(sbatch scripts/trim.sh "$R1" results/trim)
trim_id=$(echo "$submit_line" | sed 's/Submitted batch job //')
# Submit the mapping job with the condition that it only starts when the
# trimming job is done, using '--dependency=afterok:':
R1_trimmed=results/trim/$(basename "$R1")
sbatch --dependency=afterok:$trim_id scripts/map.sh "$R1_trimmed" results/map
done
# If you give the mapping and counting jobs the same name with `#SBATCH --job-name=`,
# then you can use '--dependency=singleton': the counting job will only start
# when ALL the mapping jobs are done:
sbatch --dependency=singleton scripts/count.sh results/map results/count_table.txt