Week 7 exercises

Introduction
In these exercises, you’ll practice with writing shell scripts to run bioinformatics programs, and looping over samples to run these scripts multiple times. The example program is TrimGalore, which we use in our RNA-Seq analysis workflow:
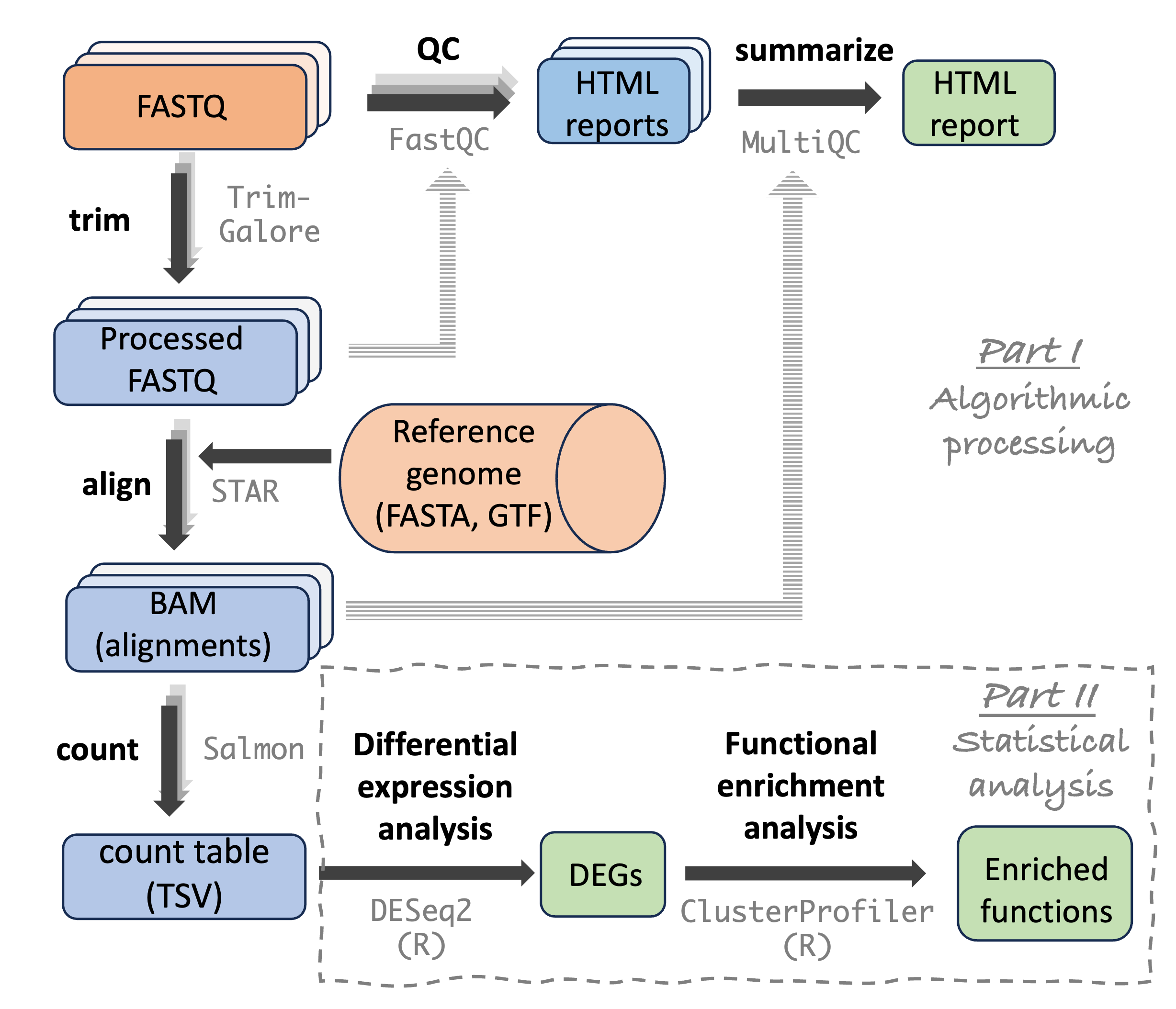
What is TrimGalore?
TrimGalore (Krueger 2021) is a tool that can trim and filter FASTQ files, removing:
- Any adapter sequences that are present in the reads1.
- Poor-quality bases at the start and end of the reads.
- Reads that have become very short after the prior two steps.
TrimGalore takes FASTQ files as input, and outputs filtered FASTQ files.
It is important to realize that when you have paired-end reads (as you do with the Garrigós et al. (2025) data), a single TrimGalore run should include both the R1 (forward read) and R2 (reverse read) file for one sample. This is because R1 and R2 files need to remain “synced”: generally, you can’t remove a read from an R1 file without also removing its mate from the R2. Operationally, this means we can’t simply run one FASTQ file at a time!
Several largely equivalent tools exist for this kind of FASTQ preprocessing — Trimmomatic and fastp are two other commonly used ones. TrimGalore itself is “just” a wrapper around yet another tool called Cutadapt, but it is simpler to use. Two advantages of TrimGalore are:
- It will auto-detect the adapters that are present in your reads.
- It can automatically run FastQC on the trimmed sequences.
Setting up
- At https://ondemand.osc.edu, start a VS Code session in
/fs/ess/PAS2880/users/$USER - Create a dir
week07/exercisesand navigate there – this should be your working dir for the exercises - Create a dir
scripts(i.e.,week07/exercises/scripts) but don’t navigate there - Create a file
README.mdfor notes and answers - You’ll be working with the
garrigos-dataFASTQ files. It’s up to you whether you use the files you already have (/fs/ess/PAS2880/users/$USER/garrigos-data), or copy these FASTQ files into adatadir withinweek07/exercises. - Optional: Create a Git repo and commit to it as appropriate while you are doing the exercises. At the end, create a GitHub repo and push your local repo to the remote, optionally tagging
@menukabhin an Issue.
Exercise 1: Get the TrimGalore container
Get the URL to the container image with the most recent TrimGalore version that you found last week in the graded assignment. Or go to https://seqera.io/containers to retrieve the link there.
Struggling to get the container link? Find it here. (Click to expand)
oras://community.wave.seqera.io/library/trim-galore:0.6.10--bc38c9238980c80eTo check that the container works, print the TrimGalore help info using the help option:
trim_galore --help. You’ll use this help info below.
Exercise 2: Write a TrimGalore script
Write a shell script scripts/trimgalore.sh that runs TrimGalore on paired-end FASTQ files as follows:
The script should run TrimGalore once for each sample’s R1-R2 pair of FASTQ files
Your script should accept and process arguments that specify (1) the input FASTQ files and (2) the output dir
Include the standard shell script header lines, various “logging statements” with
echolike we did in the FastQC script, and a line that prints the TrimGalore version.For each of the items below, figure out the relevant TrimGalore option, and use that option:
- Tell it that the reads are paired-end2.
- Set the output dir
- Make it run FastQC on the trimmed FASTQ files
What are the default thresholds for the Phred quality score and read length? Can you describe what these parameters do? (You don’t have to change them here, the defaults will work fine for us.)
Hint: See the relevant parts of the TrimGalore help pages (Click to expand)
trim_galore --help USAGE:
trim_galore [options] <filename(s)>
--paired This option performs length trimming of quality/adapter/RRBS trimmed reads for
paired-end files.
-o/--output_dir <DIR> If specified all output will be written to this directory instead of the current
directory. If the directory doesn't exist it will be created for you.
-j/--cores INT Number of cores to be used for trimming [default: 1].
--fastqc Run FastQC in the default mode on the FastQ file once trimming is complete.
--fastqc_args "<ARGS>" Passes extra arguments to FastQC.
-a/--adapter <STRING> Adapter sequence to be trimmed. If not specified explicitly, Trim Galore will
try to auto-detect whether the Illumina universal, Nextera transposase or Illumina
small RNA adapter sequence was used.
-q/--quality <INT> Trim low-quality ends from reads in addition to adapter removal. [...]
Default Phred score: 20.
--length <INT> Discard reads that became shorter than length INT because of either
quality or adapter trimming. A value of '0' effectively disables
this behaviour. Default: 20 bp.Hint: Struggling? Here is a script skeleton (Click to expand)
Replace the <...> parts with your code:
<...header-lines...>
# Constants
TRIMGALORE_CONTAINER=<...>
# Copy the automatically available variables to descriptively named ones
R1=<...>
R2=<...>
outdir=<...>
# Report
echo "# Starting script trimgalore.sh"
<...print the date and time...>
echo "# Input R1 FASTQ file: <...>"
<...print other variables...>
# Create the output dir
<...>
# Run TrimGalore
apptainer exec <...> \
trim_galore \
<...>
"$R1" \
"$R2"
# Report
echo
echo "# TrimGalore version:"
<...>
<...other final reporting...>--cores option for now (Click to expand)
Since you’re running the program interactively on your single-core VS Code compute job, you only have 1 core at your disposal — and that’s TrimGalore’s default. Therefore, don’t adjust this setting here. Next week, you’ll submit this script as a batch job, giving you the opportunity to use multiple cores.
Exercise 3: Run your TrimGalore script once
Run your TrimGalore script for the FASTQ files of the
ERR10802863sample. (Run it directly usingbash <script-path> ..., don’t submit it as a batch job.)Check if everything went well by scanning the output that is printed to screen, and by taking a look at TrimGalore’s output files. What outputs are you finding? (If something went wrong, go back to your
trimgalore.shscript and try to fix it, and describe your process in the README.)Take a closer look at the output that TrimGalore printed to screen (note that most of that is also saved in the
*_trimming_report.txtfile in the output dir):- In each file (R1 and R2), in what percentage of reads were adapters detected?
- In each file (R1 and R2), what percentage of bases were quality trimmed?
- What percentage of reads were removed because they were too short?
Download the FastQC output for the trimmed R1 file and compare it to the output file for last week’s FastQC run on the raw R1 file. E.g.: can you see any differences in overall sequence quality, and do the very short reads with only Ns appear to have been removed?
Exercise 4: Run your TrimGalore script for multiple samples
In the README, write and run a
forloop that will run your TrimGalore script for FASTQ files from multiple samples. Because you aren’t yet submitting batch jobs (you’ll do that next week!), the script will run consecutively for each sample, so this will take a while. Therefore, only loop over the 7 sample IDs that start withERR1080286. (Make sure you are still running the script once per sample!)Monitor the output printed to screen, and check TrimGalore’s output files once it’s done.
Solutions
Exercise 2
The TrimGalore options you were looking for (Click to expand)
--pairedto indicate that the FASTQ files are paired-end.--output_dir(or equivalently,-o) to specify the output directory.--fastqcto run FastQC on the trimmed FASTQ files.
A TrimGalore shell script (Click to expand)
#!/bin/bash
set -euo pipefail
# Constants
TRIMGALORE_CONTAINER=oras://community.wave.seqera.io/library/trim-galore:0.6.10--bc38c9238980c80e
# Copy the placeholder variables
R1="$1"
R2="$2"
outdir="$3"
# Report
echo "# Starting script trimgalore.sh"
date
echo "# Input R1 FASTQ file: $R1"
echo "# Input R2 FASTQ file: $R2"
echo "# Output dir: $outdir"
echo
# Create the output dir
mkdir -p "$outdir"
# Run TrimGalore
apptainer exec "$TRIMGALORE_CONTAINER" \
trim_galore \
--paired \
--fastqc \
--output_dir "$outdir" \
"$R1" \
"$R2"
# Report
echo
echo "# TrimGalore version:"
apptainer exec "$TRIMGALORE_CONTAINER" \
trim_galore -v
echo "# Successfully finished script trimgalore.sh"
dateQuality and length threshold options & interpretation (Click to expand)
The option
-q(short notation) or--quality(long notation) can be used to adjust the base quality score threshold, which has the Phred unit we’ve discussed. The default threshold is 20, which corresponds to an estimated 0.01 error rate. TrimGalore will trim bases with a lower quality score than this threshold from the ends of the reads. It will not remove or mask bases with lower quality scores elsewhere in the read.The option
-l(short notation) or--length(long notation) can be used to adjust the read length threshold: any reads that are shorter than this after adapter and quality trimming will be removed. The default threshold is 20 bp. (When the input is paired-end, the corresponding second read will also be removed, regardless of its length: paired-end FASTQ files always need to contain both the R1 and R2 for each pair; orphan reads would need to be stored in a separate file.)
Exercise 3
1 - Run the script (Click to expand)
# Exercise 2
R1=../garrigos-data/fastq/ERR10802863_R1.fastq.gz
R2=../garrigos-data/fastq/ERR10802863_R2.fastq.gz
bash scripts/trimgalore.sh "$R1" "$R2" results/trimgalore# Starting script trimgalore.sh
Thu Mar 28 10:34:24 EDT 2024
# Input R1 FASTQ file: ../garrigos-data/fastq/ERR10802863_R1.fastq.gz
# Input R2 FASTQ file: ../garrigos-data/fastq/ERR10802863_R2.fastq.gz
# Output dir: results/trimgalore
Multicore support not enabled. Proceeding with single-core trimming.
Path to Cutadapt set as: 'cutadapt' (default)
Cutadapt seems to be working fine (tested command 'cutadapt --version')
Cutadapt version: 4.4
# [...output truncated...]2 - Check the output files (Click to expand)
ls -lh results/trimgaloretotal 42M
-rw-rw----+ 1 jelmer PAS0471 2.4K Mar 28 10:49 ERR10802863_R1.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 674K Mar 28 10:49 ERR10802863_R1_val_1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 349K Mar 28 10:49 ERR10802863_R1_val_1_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 20M Mar 28 10:49 ERR10802863_R1_val_1.fq.gz
-rw-rw----+ 1 jelmer PAS0471 2.3K Mar 28 10:49 ERR10802863_R2.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 676K Mar 28 10:50 ERR10802863_R2_val_2_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 341K Mar 28 10:50 ERR10802863_R2_val_2_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 21M Mar 28 10:49 ERR10802863_R2_val_2.fq.gz3 - Check the trimming results (Click to expand)
Note that the adapter- and quality trimming results summary for the R1 and R2 files are separated widely among all the output that TrimGalore prints — they are printed below (first for the R1, then for the R2 file), and the answers are:
- 13.0% (R1) and 10.1% (R2) of reads had adapters
- 3.6% (R1) and 3.5% (R2) of bases were quality-trimmed
=== Summary === Total reads processed: 500,000 Reads with adapters: 65,070 (13.0%) Reads written (passing filters): 500,000 (100.0%) Total basepairs processed: 35,498,138 bp Quality-trimmed: 1,274,150 bp (3.6%) Total written (filtered): 34,138,461 bp (96.2%)=== Summary === Total reads processed: 500,000 Reads with adapters: 50,357 (10.1%) Reads written (passing filters): 500,000 (100.0%) Total basepairs processed: 36,784,563 bp Quality-trimmed: 1,283,793 bp (3.5%) Total written (filtered): 35,440,230 bp (96.3%)Just before the FastQC logs is a line that reports how many reads were removed due to the length filter — the answer is 6.79%.
Number of sequence pairs removed because at least one read was shorter than the length cutoff (20 bp): 33955 (6.79%)
4 - Check the FastQC output HTML file (Click to expand)
Yes, there are clear differences in:
The mean quality scores, as shown by both the “Per base sequence quality” and the “Per sequence quality scores” graphs => qualities are higher after trimmming
The “Per sequence GC content”, “Sequence Length Distribution”, and “Overrepresented sequences” graphs, all of which had artefacts related to those very short reads that only contained Ns: these reads have been removed by the trimming process.
Exercise 4
1 - Run the script for multiple samples (Click to expand)
Test the loop:
for R1 in ../garrigos-data/fastq/ERR1080286*_R1.fastq.gz; do
R2=${R1/_R1/_R2}
echo bash scripts/trimgalore.sh "$R1" "$R2" results/trimgalore
donebash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802863_R1.fastq.gz ../garrigos-data/fastq/ERR10802863_R2.fastq.gz results/trimgalore
bash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802864_R1.fastq.gz ../garrigos-data/fastq/ERR10802864_R2.fastq.gz results/trimgalore
bash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802865_R1.fastq.gz ../garrigos-data/fastq/ERR10802865_R2.fastq.gz results/trimgalore
bash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802866_R1.fastq.gz ../garrigos-data/fastq/ERR10802866_R2.fastq.gz results/trimgalore
bash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802867_R1.fastq.gz ../garrigos-data/fastq/ERR10802867_R2.fastq.gz results/trimgalore
bash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802868_R1.fastq.gz ../garrigos-data/fastq/ERR10802868_R2.fastq.gz results/trimgalore
bash scripts/trimgalore.sh ../garrigos-data/fastq/ERR10802869_R1.fastq.gz ../garrigos-data/fastq/ERR10802869_R2.fastq.gz results/trimgaloreLooks good? Then run the loop:
for R1 in ../garrigos-data/fastq/ERR1080286*_R1.fastq.gz; do
R2=${R1/_R1/_R2}
bash scripts/trimgalore.sh "$R1" "$R2" results/trimgalore
done# (Output not shown, same as earlier but should repeat for each sample)2 - Check the output files (Click to expand)
ls -lh results/trimgaloretotal 949M
-rw-rw----+ 1 jelmer PAS0471 20M Mar 28 11:18 ERR10802863_R1_val_1_.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.4K Mar 28 11:17 ERR10802863_R1.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 674K Mar 28 11:18 ERR10802863_R1_val_1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 349K Mar 28 11:18 ERR10802863_R1_val_1_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 21M Mar 28 11:18 ERR10802863_R2_val_2_.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.3K Mar 28 11:18 ERR10802863_R2.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 676K Mar 28 11:18 ERR10802863_R2_val_2_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 341K Mar 28 11:18 ERR10802863_R2_val_2_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 21M Mar 28 11:19 ERR10802864_R1_val_1_.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.7K Mar 28 11:18 ERR10802864_R1.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 675K Mar 28 11:19 ERR10802864_R1_val_1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 349K Mar 28 11:19 ERR10802864_R1_val_1_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 22M Mar 28 11:19 ERR10802864_R2_val_2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.6K Mar 28 11:19 ERR10802864_R2.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 671K Mar 28 11:19 ERR10802864_R2_val_2_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 336K Mar 28 11:19 ERR10802864_R2_val_2_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 21M Mar 28 11:20 ERR10802865_R1_val_1_.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.6K Mar 28 11:19 ERR10802865_R1.fastq.gz_trimming_report.txt
-rw-rw----+ 1 jelmer PAS0471 682K Mar 28 11:20 ERR10802865_R1_val_1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 361K Mar 28 11:20 ERR10802865_R1_val_1_fastqc.zip
[...output truncated...]