Slurm batch jobs – part II
Week 7 – Lecture B

1 Introduction
1.1 Learning goals
Now that you know the basics of submitting and managing Slurm batch jobs, you will learn:
- The most common Slurm/
sbatchoptions, including those to request:- More time, cores, and memory for jobs
- An email notification when jobs e.g. fail or finish
- How to run Slurm jobs in practice, including how to submit many jobs at the same time
- How to make sure that your jobs succeeded
1.2 Getting ready
- At https://ondemand.osc.edu, start a VS Code session in
/fs/ess/PAS2880/users/$USER - In the terminal, navigate to your
week07dir
1.3 Testing the Shellcheck extension
- Create a new script
scripts/shellcheck-test.shand open it. - If Shellcheck works, you should immediately see a small red squiggly line on the first line – and when you hover over that, the following warning:
Below: Hover over the squiggly line to see what this warning is about.
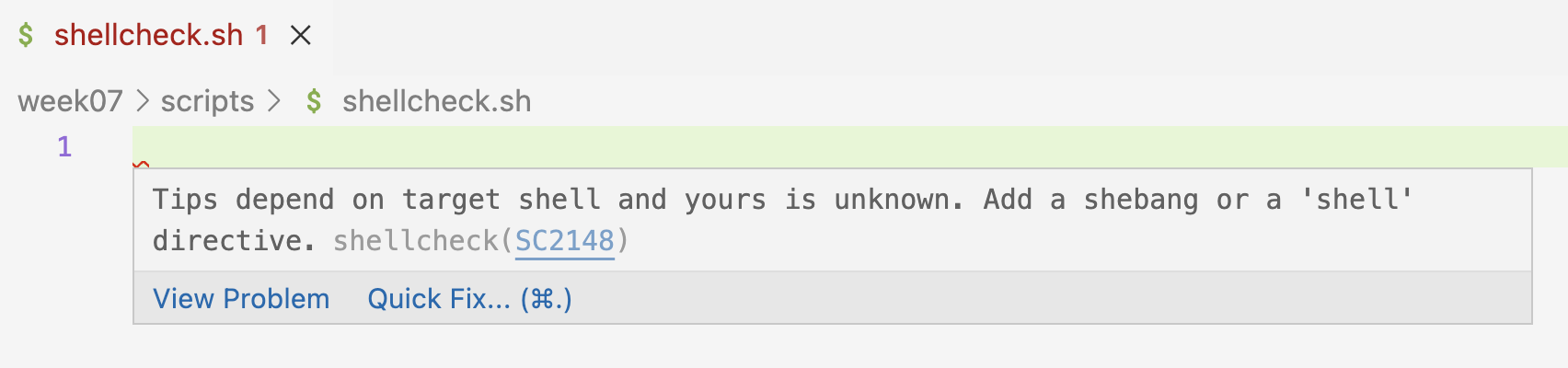
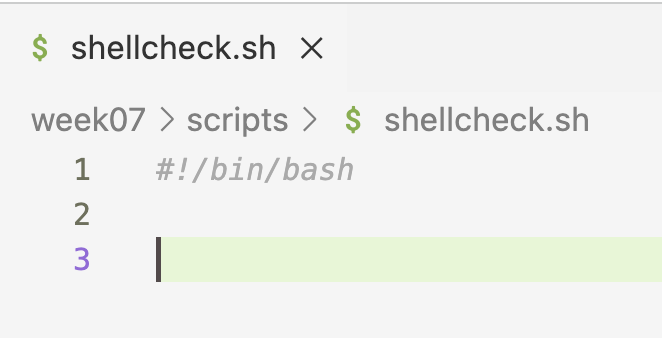
- So, Shellcheck wants you to add a shebang line, and once you do so, the squiggly line disappears:
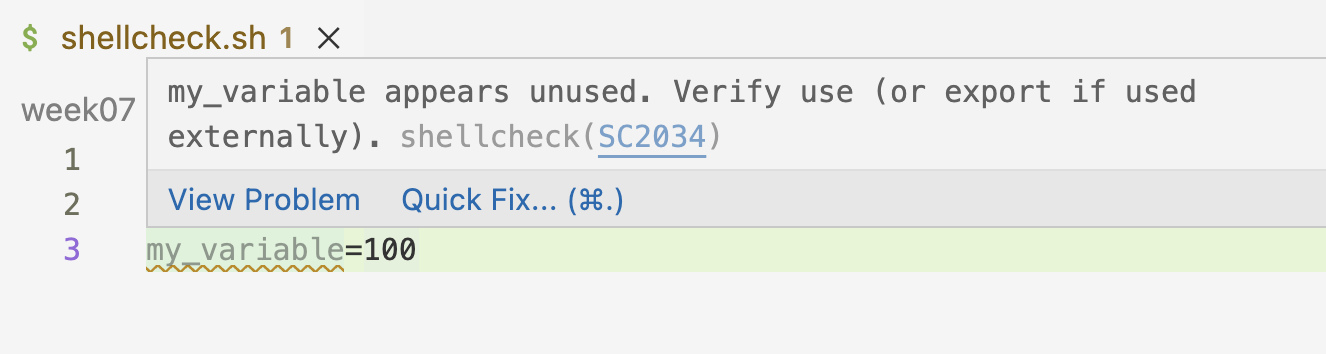
- That was one example of a handy Shellcheck tip. Another is when you have assigned a variable, but don’t use (reference it):

Below: Hovering over the line again tells you the reason for the warning.
- Referencing the variable in question makes the squiggly line disappear:
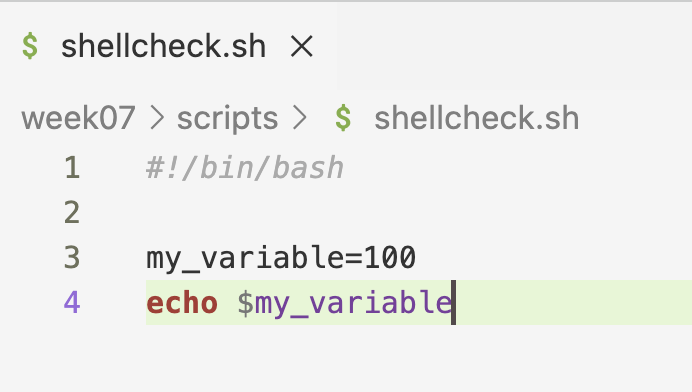
- Conversely, when you reference a variable that hasn’t been assigned:
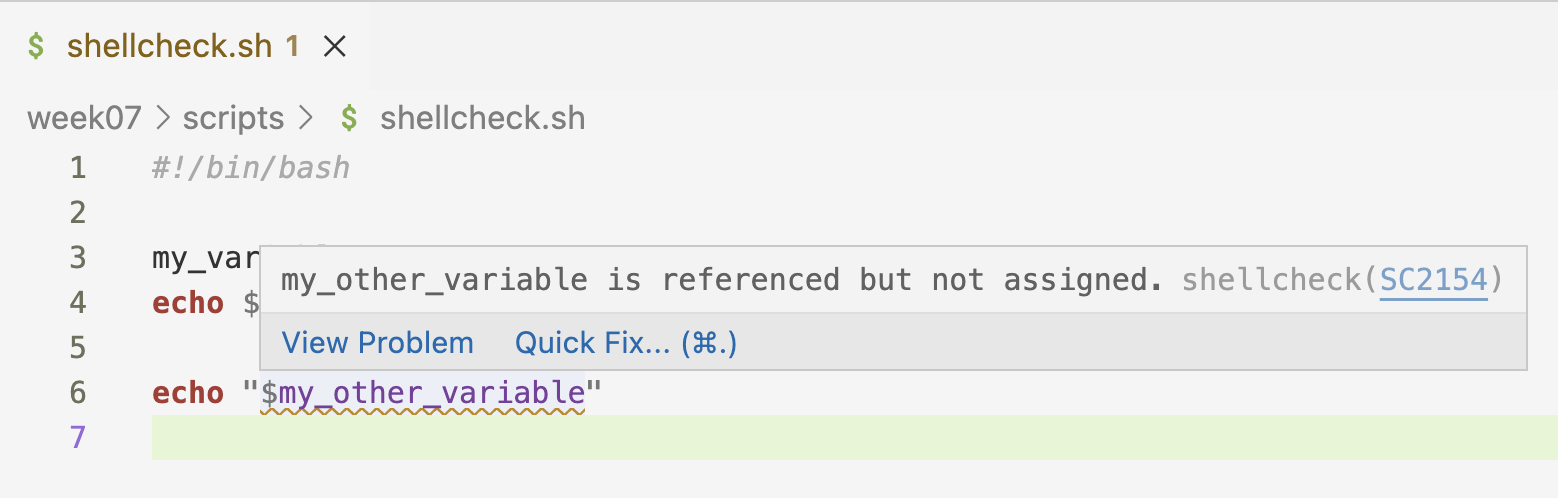
- These tips can be extremely useful! Also in cases where you e.g. make a typo in a variable name, or renamed a variable but didn’t do so consistently.
2 Common Slurm/sbatch options
Here, we’ll go through the most commonly used sbatch options (except --account, which was covered in the previous lecture). As pointed out above, each of these options can be:
- passed on the command line:
sbatch --account=PAS2880 myscript.sh(has precedence over the next) and/or - added at the top of the script you’re submitting:
#SBATCH --account=PAS2880.
Also, note that many options have a corresponding long (e.g. --account=) and short format (e.g -A). For clarity, we’ll stick to long-format options here.
2.1 Time with --time
If you suspect or know that the default time limit of 1 hour may not be enough for your job, use the --time option to request more time. Note that tour job will be killed (stopped) as soon as it hits the time limit!
For single-node jobs, up to 168 hours / 7 days can be requested. (If that’s not enough, you can request access to a separate queue for long jobs of up to 14 days, called the longserial queue.)
OSC bills projects for the time that jobs actually used, not what you reserved. And recall that batch jobs will stop as soon as the focal script has finished running. But before you decide to ask for 168 hours for every job, note that jobs that request more time are likely to have to wait longer before they start.
When you reserve an amount of time with --time, this is the “wall time”. What does wall time mean? This is simply the actual amount of time that passes on the (wall) clock. This term is used to contrast it with “core hours”, which is the number of hours multiplied by the number of cores:
| # hours | # cores | wall time | # core hours |
|---|---|---|---|
| 2 | 1 | 2 | 2 |
| 2 | 8 | 2 | 16 |
A number of formats can be used to specify the time, including:
| Format | Example | Meaning | |
|---|---|---|---|
| minutes | 60 |
60 minutes | |
| hours:minutes:seconds | 1:00:00 |
60 minutes | |
| days-hours | 2-12 |
two-and-a-half days (60 hours) |
For example, requesting 2 hours in the “minute-format” inside a shell script:
#!/bin/bash
#SBATCH --time=120It is common to be uncertain about how much time your job will take (i.e., how long it will take for your script to finish). Whenever this happens, ask for more, perhaps much more, time than what you think/guesstimate you will need. It is really annoying to have a job run out of time after several hours, while the increase in queueing time for jobs that request more time is often quite minimal at OSC.
Exercise: exceed the time limit
Modify the sleep.sh script to reserve only 1 minute for the job, while making the script run for longer than that by increasing the sleep sleeping time to at least 100 seconds.
If you succeed in exceeding the time limit, an error message will be printed. Where do you think this error message will be printed: to the screen, in the Slurm log file, or in sleep.txt?
After waiting for the job to be killed after 60 seconds, check if you were correct and what the error message is.
2.2 Cores with --cpus-per-task
First, note that Slurm mostly uses the terms “core” and “CPU” interchangeably1. More generally with bioinformatics tools, “thread” is also commonly used interchangeably with core/CPU2. Therefore, for our purposes, you can think of “core”, “CPU”, and “thread” as synonyms that all refer to the same sub-parts/components of a node that you can reserve and use separately.
Running a bioinformatics program with multiple threads/cores/CPUs (“multi-threading”) is very common, and this can make the running time of such programs much shorter. While the specifics depend on the program, using 8-12 cores is often a sweet spot, whereas asking for even more cores can lead to rapidly diminishing returns.
While there are multiple relevant Slurm options (see the box below), I recommend that you specify the number of threads/cores/CPUs with the option --cpus-per-task (-c).
Typically, inside your shell script, you will also want to inform the program that your script runs about the number of available cores – most have an option like --cores or --threads. You should set this to the same value n as the --cpus-per-task. An example, where we ask for 8 CPUs/cores/threads:
#!/bin/bash
#SBATCH --cpus-per-task=8
# And we tell a fictional program about that number of cores:
trimming_program.py --cores 8 sampleA_R1.fastq.gzYou may have noticed that the long form of the above option is --cpus-per-task instead of just, say, --cpus. What is that about? Let’s go over nodes and tasks:
Tasks are independent processes that a computer can run. And recall that nodes are the large computers that a supercomputer is made of.
In bioinformatics, running multiple processes (tasks) or needing multiple nodes in a single batch job is not common.
You can specify the number of nodes with
--nodesand the number of tasks with--ntasksand/or--ntasks-per-node; all have defaults of 1 (see the table below).Unless you have very clear reasons to, I recommend not to use the abovementioned options to change the number of tasks or nodes, so your job will have 1 of each. When you do this, the above
-coption simply specifies the number of cores.For example, only ask for more than one node when a program is parallelized with e.g. “MPI”, which is rare in bioinformatics.
For jobs that require multiple processes (tasks), you can use
--ntasks=nor--ntasks-per-node=n— this is also quite rare! However, note in practice, specifying the number of tasksnwith one of these options is equivalent to using--cpus-per-task=n, in the sense that both ask forncores that can subsequently be used by a program in your script. Therefore, some people usetasksas opposed tocpusfor multi-threading, and you may run across this in the OSC documentation too. (Yes, this is confusing!)
Here is an overview of the options related to cores, tasks, and nodes:
| Resource/use | short | long | default |
|---|---|---|---|
| Nr. of cores/CPUs/threads (per task) | -c 1 |
--cpus-per-task=1 |
1 |
| Nr. of “tasks” (processes) | -n 1 |
--ntasks=1 |
1 |
| Nr. of tasks per node | - | --ntasks-per-node=1 |
1 |
| Nr. of nodes | -N 1 |
--nodes=1 |
1 |
2.3 RAM memory with --mem
The --mem option specifies the maximum amount of RAM (Random Access Memory) available to your job.
The default amount of memory will depend on how many cores you reserved. Standard cores have 4 GB of memory “on it”, and therefore, the default amount of memory you will get is 4 GB is per reserved core. For example, if you specify --cpus-per-task=4, you will have 16 GB of memory. And the default number of cores is 1, which gives you 4 GB.
Like with the time limit, your job gets canceled by Slurm when it hits the memory limit (see the box below).
Because it is common to ask for multiple cores and due to the above-mentioned adjustment of the memory based on the number of cores, you will often end up having enough memory automatically, and won’t need to use the --mem option.
The maximum amount of memory you can request on regular Pitzer compute nodes is 178 GB. If you need more than that, you should use one of the specialized largemem or hugemem nodes — switching to such a node generally happens automatically based on your requested amount3 — see this OSC page for details on node types and associated memory on Pitzer.
The default --mem unit is MB (MegaBytes); append G for GB (i.e. 100 means 100 MB, 10G means 10 GB). For example, to request 20 GB of RAM:
#!/bin/bash
#SBATCH --mem=20GWhereas you get a very clear Slurm error message when you hit the time limit (as seen in the exercise above), hitting the memory limit can result in a variety of errors.
But look for keywords such as “Killed”, “Out of Memory” / “OOM”, and “Core Dumped”, as well as actual “dumped cores” in your working dir (large files with names like core.<number>, these can be deleted).
If you see these or otherwise suspect that your job ran out of memory, resubmit it with an increased requested amount of memory.
To request that a bach job runs on a non-default node type, you should use the --partition option. For example:
#!/bin/bash
#SBATCH --partition=hugemem
#SBATCH --mem=900GExercise: Adjusting cores and memory
Think about submitting a shell script that runs a bioinformatics tool like FastQC as a batch job, in the following two scenarios:
The program has an option
--cores, and you want to set that to 12. The program also says you’ll need 60 GB of memory. Which relevant#SBATCHoption values will you use?The program has an option
--threads, and you want to set that to 8. The program also says you’ll need 25 GB of memory. Which relevant#SBATCHoption values will you use?
2.4 Slurm log file names with --output
As you saw above, output from a script that would normally be printed to screen will end up in a Slurm log file when it is submitted as a batch job. By default, this file is created in the directory from which you submitted the script, and is called slurm-<job-number>.out, e.g. slurm-12431942.out.
But it is possible to change the name of this file. For instance, it can be useful to include the name of the bioinformatics program that the script runs (or another kind of keyword describing the function of the script), so that it’s easier to recognize this file later. You can do this with the --output option, e.g. --output=slurm-fastqc.out if you were running FastQC.
But you’ll generally want to keep the batch job number in the file name too4. Since you won’t know the batch job number in advance, you need a trick here – and that is to use %j, which represents the batch job number:
#!/bin/bash
#SBATCH --output=slurm-fastqc-%j.outstdout and stderr, and separating them
The output that programs (including basic Unix commands) print to screen can be divided into two types:
- Standard output (
stdout) for regular output - Standard error (
stderr) for error messages and sometimes warnings messages
Most of the time, it’s not obvious that these are two separate types, but it possible to treat them separately. For example, while both stdout and stderr by default end up in the same Slurm log file, it is possible to separate them into different files. You can do that by using the --error option, e.g.:
#SBATCH --output=slurm-fastqc-%j.out
#SBATCH --error=slurm-fastqc-%j.errIn the above example, only stdout will go into the file specified with --output, and stderr will go into the file specified with --error. Separating stdout and stderr like that could be useful because it may make it easier to spot errors, and an empty --error file would mean that there were no errors.
However, reality is more messy: some programs print their main output (results) not to a file but to standard out, in which case all logging output, i.e. errors and regular messages alike, is designated as standard error. And some other programs use stdout for all messages. Therefore, with Slurm, I personally only specify --output, such that both stdout and stderr end up in that file.
2.5 Notification emails with --mail-type
You can use the --mail-type option to receive emails when a job begins, completes successfully, or fails. You don’t have to specify your email address: you’ll be automatically emailed on the address linked to your OSC account. I would recommend that for…
Shorter-running jobs (roughly up to a few hours): only have Slurm send emails if they fail (
--mail-type=FAIL). Especially when you submit many jobs at a time, you may otherwise overlook failure for some jobs.#!/bin/bash #SBATCH --mail-type=FAILLonger-running jobs, have Slurm send emails both for failed and successfully completed jobs (
--mail-type=END,FAIL). This is helpful because you don’t want to have to keep checking in on jobs that run for many hours5.#!/bin/bash #SBATCH --mail-type=END,FAIL
You may also find the --mail-type values TIME_LIMIT_90, TIME_LIMIT_80, and TIME_LIMIT_50 useful for very long-running jobs, which will warn you when the job is at 90/80/50% of the time limit. For example, it is possible to email OSC to ask for an extension on individual jobs. You shouldn’t do this often, but if you have a job that ran for 6 days and it looks like it may time out, this may well be worth it.
2.6 Table with sbatch options
Here are the main options we discussed above:
| Resource/use | long option example | short option | default |
|---|---|---|---|
| Project to be billed | --account=PAS2880 |
-A |
N/A |
| Time limit | --time=4:00:00 |
-t |
1:00:00 |
| Nr of cores | --cpus-per-task=1 |
-c |
1 |
| Memory limit per node | --mem=4G |
- | 4G |
| Log output file | --output=slurm-fastqc-%j.out |
-o |
|
| Put error output (stderr) from into a separate file | --error=slurm-fastqc-%j.err |
||
| Get email when job ends / ends or fails |
--mail-type=END --mail-type=END,FAIL |
- |
And some additional ones:
| Resource/use | long option example |
|---|---|
Job name (will e.g. be displayed in squeue output) |
--job-name=fastqc |
| Partition (=queue type) | --partition=longserial --partition=hugemem |
| Only allow job to begin after a specific time | --begin=2025-04-05T12:00:00 |
| Only allow job to begin after another job has successfully finished | --dependency=afterok:123456 |
| Nr of nodes | --nodes=1 |
| Nr of “tasks” (processes) | --ntasks=1 |
| Nr of tasks per node | --ntasks-per-node=1 |
3 Batch job examples with FastQC
Last week, we used a for loop to run FastQC interactively for every FASTQ file. This ran quickly, but keep in mind that our practice FASTQ files are only 5% of their original sizes. Additionally, other bioinformatics programs may take much longer. To speed things up, and not block the terminal while programs are running, we will submit a batch job for each sample/file, so that many jobs can run simultaneously6.
So, let’s practice with submitting a batch job for each FastQC run. We’ll first submit a job for a single file, and then loop over all files to submit many jobs. You should have the FastQC script in scripts/fastqc.sh – if not, see the box below.
#!/bin/bash
set -euo pipefail
# Load the OSC module for FastQC
module load fastqc/0.12.1
# Copy the placeholder variables
fastq="$1"
outdir="$2"
# Initial logging
echo "# Starting script fastqc.sh"
date
echo "# Input FASTQ file: $fastq"
echo "# Output dir: $outdir"
echo
# Create the output dir if needed
mkdir -p "$outdir"
# Run FastQC
fastqc --outdir "$outdir" "$fastq"
# Final logging
echo
echo "# Used FastQC version:"
fastqc --version
echo
echo "# Successfully finished script fastqc.sh"
date3.1 Modify the script for usage with Slurm
Next, add Slurm options with the following #SBATCH lines in the script – note that you’ll ask for 4 cores:
#!/bin/bash
#SBATCH --account=PAS2880
#SBATCH --cpus-per-task=4
#SBATCH --mail-type=FAIL
#SBATCH --output=slurm-fastqc-%j.outWhen you have multiple cores at your disposal in a batch job, you should almost always explicitly tell the program that you’re running about this. Some programs default to using 1 core while others try to auto-detect the number of cores, but any auto-detection tends to be inappropriate: the program would detect the total number of cores on the OSC node rather than the number that we have reserved.
Exercise: Let FastQC use multiple cores
Change the FastQC command in the script so that FastQC will use all 4 reserved cores. Run fastqc --help to find the relevant option.
Click to see the solution
The relevant section of the Help text:
-t --threads Specifies the number of files which can be processed
simultaneously. Each thread will be allocated 250MB of
memory so you shouldn't run more threads than your
available memory will cope with, and not more than
6 threads on a 32 bit machineLet FastQC know how many cores you have reserved by editing the command to include --threads 4:
fastqc --threads 4 --outdir "$outdir" "$fastq"3.2 Submit a single test batch job
Now, you’re ready to submit the script. Let’s test it for one sample first:
# We'll assign the FASTQ file to a variable first
# (e.g. to shorten the next sbatch command):
fastq_file=../garrigos-data/fastq/ERR10802863_R1.fastq.gz
ls -lh "$fastq_file"-rw-rw----+ 1 jelmer PAS0471 21M Sep 9 13:46 sbatch scripts/fastqc.sh "$fastq_file" results/fastqcSubmitted batch job 37615710Monitor the batch job
Next, monitor your job by checking its status in the Slurm queue:
squeue -u $USER -lTue Sep 30 09:21:48 2025
JOBID PARTITION NAME USER STATE TIME TIME_LIMI NODES NODELIST(REASON)
37615710 cpu-exp fastqc.s jelmer RUNNING 0:01 1:00:00 1 p0510After a while:
- You will know that your job has stopped running (either due to failure or because it finished succesfully) when it is no longer listed in the
squeueoutput - If you additionally didn’t receive an email from Slurm, this implies the job succeeded – job failure should have triggered an email due to the
#SBATCH --mail-type=FAILline.
Check the output files
Even if you’ve inferred that your job succeeded from the abovementioned clues, don’t forget to look at the output files to confirm this:
Slurm log file: The combination of having strict bash
setsettings and the “Succesfully finished”echoline at the end of the script means that if you see that line printed, the script encountered no errors. Nevertheless, you should also look at the rest of this file, because a program’s “logging statements” may have useful information, including output summaries but also warnings.The program’s main output files: As discussed, most bioinformatics tools don’t just print “logging” information that ends up in the Slurm log file, but will also have output files (in the case of of FastQC, and HTML and a Zip file). Therefore, always check the output dir for the presence of such output files.
Checking the Slurm log file:
Make sure it’s there:
lsslurm-fastqc-37615710.outQuick check that the “marker” line that indicates a successful run is printed:
tail slurm-fastqc*.out==> slurm-fastqc-37615710.out <== Approx 90% complete for ERR10802863_R1.fastq.gz Approx 95% complete for ERR10802863_R1.fastq.gz Approx 100% complete for ERR10802863_R1.fastq.gz Analysis complete for ERR10802863_R1.fastq.gz # Used FastQC version: FastQC v0.12.1 # Successfully finished script fastqc.sh Tue Sep 30 09:21:51 AM EDT 2025Take a closer look at the file:
less slurm-fastqc* # [output not shown]
Finally, check for the presence of the main FastQC output files:
ls -lh results/fastqctotal 1.1M
-rw-rw----+ 1 jelmer PAS0471 718K Sep 30 09:21 ERR10802863_R1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 364K Sep 30 09:21 ERR10802863_R1_fastqc.zipIt can also be a good idea to pay attention to the sizes of output files, as shown by ls -lh. Especially if you’re not going to open the output files right away, this can be yet another clue as to whether the program succeeded. When they fail, some programs programs produce empty or extremely small output files – their presence should always be a red flag.
Clean up
Because this was a test-run, and you will now run a loop to analyze each sample, let’s remove the outputs:
rm -r results/fastqc slurm-fastqc*Exercise: Trigger a job failure email
Submit the script
scripts/fastqc.shas a batch job again, but don’t give the script any arguments when you do so.This will make the job fail, so wait for a failure email to arrive. (If it doesn’t arrive immediately, check the queue to make sure your job isn’t still pending.)
Optional: if you prefer, set a “Rule” in Outlook so the Slurm emails will not go to your Inbox but to a specific folder for such emails.
3.3 Submit a job for each file
Next, we’ll loop over all FASTQ files to submit a job for each file. Though before you do so, let’s do a “mock-run” – it your preface the command with echo, the command will just be printed instead of executed. That can be really helpful with loops, e.g. to make sure you are looping over the files you want:
for fastq_file in ../garrigos-data/fastq/*fastq.gz; do
echo sbatch scripts/fastqc.sh "$fastq_file" results/fastqc
donesbatch scripts/fastqc.sh ../garrigos-data/fastq/ERR10802863_R1.fastq.gz results/fastqc
sbatch scripts/fastqc.sh ../garrigos-data/fastq/ERR10802863_R2.fastq.gz results/fastqc
sbatch scripts/fastqc.sh ../garrigos-data/fastq/ERR10802864_R1.fastq.gz results/fastqc
sbatch scripts/fastqc.sh ../garrigos-data/fastq/ERR10802864_R2.fastq.gz results/fastqc
sbatch scripts/fastqc.sh ../garrigos-data/fastq/ERR10802865_R1.fastq.gz results/fastqc
sbatch scripts/fastqc.sh ../garrigos-data/fastq/ERR10802865_R2.fastq.gz results/fastqc
# [...output truncated...]That looked as expected, so now you can actually submit the batch jobs:
for fastq_file in ../garrigos-data/fastq/*fastq.gz; do
sbatch scripts/fastqc.sh "$fastq_file" results/fastqc
doneSubmitted batch job 37615718
Submitted batch job 37615719
Submitted batch job 37615720
Submitted batch job 37615721
Submitted batch job 37615722
Submitted batch job 37615723
# [...output truncated...]All these jobs were submitted in very quick succession, and we got our prompt back. Check the queue:
squeue -u $USER -lTue Sep 30 06:36:01 2025
JOBID PARTITION NAME USER STATE TIME TIME_LIMI NODES NODELIST(REASON)
37615718 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615719 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615720 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615721 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615722 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615723 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615724 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
37615725 cpu-exp fastqc.s jelmer RUNNING 0:03 1:00:00 1 p0840
# [...output truncated...] Nice! At least in this case, all jobs started running simultaneously right away! Once all jobs have finished (are no longer listed in squeue output), you should have many Slurm log files:
lsresults slurm-fastqc-37615719.out slurm-fastqc-37615723.out slurm-fastqc-37615727.out slurm-fastqc-37615731.out slurm-fastqc-37615735.out
scripts slurm-fastqc-37615720.out slurm-fastqc-37615724.out slurm-fastqc-37615728.out slurm-fastqc-37615732.out slurm-fastqc-37615736.out
slurm-fastqc-37615717.out slurm-fastqc-37615721.out slurm-fastqc-37615725.out slurm-fastqc-37615729.out slurm-fastqc-37615733.out slurm-fastqc-37615737.out
slurm-fastqc-37615718.out slurm-fastqc-37615722.out slurm-fastqc-37615726.out slurm-fastqc-37615730.out slurm-fastqc-37615734.out slurm-fastqc-37615738.outYou now arguably have too many Slurm log files to go through, so what to do next? Here’s what I would generally recommend:
Take a close look at (at least) one Slurm log file:
less slurm-fastqc-* # [output not shown]Check the last few lines of all of them:
tail slurm-fastqc-*==> slurm-fastqc-37615717.out <== Approx 85% complete for ERR10802863_R1.fastq.gz Approx 90% complete for ERR10802863_R1.fastq.gz Approx 95% complete for ERR10802863_R1.fastq.gz Approx 100% complete for ERR10802863_R1.fastq.gz Analysis complete for ERR10802863_R1.fastq.gz # Used FastQC version: FastQC v0.12.1 # Successfully finished script fastqc.sh Tue Sep 30 09:35:55 AM EDT 2025 ==> slurm-fastqc-37615718.out <== Approx 85% complete for ERR10802864_R1.fastq.gz Approx 90% complete for ERR10802864_R1.fastq.gz Approx 95% complete for ERR10802864_R1.fastq.gz Approx 100% complete for ERR10802864_R1.fastq.gz Analysis complete for ERR10802864_R1.fastq.gz # Used FastQC version: FastQC v0.12.1 # Successfully finished script fastqc.sh Tue Sep 30 09:36:03 AM EDT 2025 # [...output truncated...]Check for the presence of the FastQC output files:
ls -lh results/fastqctotal 25M -rw-rw----+ 1 jelmer PAS0471 718K Sep 30 06:35 ERR10802863_R1_fastqc.html -rw-rw----+ 1 jelmer PAS0471 364K Sep 30 06:35 ERR10802863_R1_fastqc.zip -rw-rw----+ 1 jelmer PAS0471 714K Sep 30 06:36 ERR10802864_R1_fastqc.html -rw-rw----+ 1 jelmer PAS0471 366K Sep 30 06:36 ERR10802864_R1_fastqc.zip -rw-rw----+ 1 jelmer PAS0471 713K Sep 30 06:36 ERR10802865_R1_fastqc.html -rw-rw----+ 1 jelmer PAS0471 367K Sep 30 06:36 ERR10802865_R1_fastqc.zip -rw-rw----+ 1 jelmer PAS0471 718K Sep 30 06:36 ERR10802866_R1_fastqc.html -rw-rw----+ 1 jelmer PAS0471 372K Sep 30 06:36 ERR10802866_R1_fastqc.zip -rw-rw----+ 1 jelmer PAS0471 710K Sep 30 06:36 ERR10802867_R1_fastqc.html # [...output truncated...]
If everything looks good, and you are convinced that all jobs have finished succesfully, all that is left is to clean up the Slurm log files. In this case, it does makes sense to keep these files around somewhere, as they contain information about our runs (when, which files, what version, etc.). But we want to move them out of our working dir. It often makes sense to store them in a subdir of the program’s outdir, e.g.:
mkdir results/fastqc/logs
mv slurm-fastqc-* logs/3.4 Recap: Making sure your jobs ran successfully
To recap, here is an overview of basic strategies to monitor your batch jobs:
To see whether your job(s) have started, check the queue (with
squeue) and/or check for Slurm log files (withls).Once the jobs are no longer listed in the queue, they will have finished: either successfully or because of an error.
When you’ve submitted many jobs that run the same script for different samples/files:
- For at least 1 one of the jobs, carefully read the full Slurm log file, and check other output files,
- Check whether no jobs have failed: via email when using
--mail-type=END, and/or by checking thetailof each log for messages like “Successfully finished script”. - Check that you have the expected number of output files and that no files have size zero (run
ls -lh).
Text will be added to the Slurm log file in real time as the running script (or the program ran by the script) outputs it. However, the output printed by commands like cat and less print are static and not updated in real-time.
Therefore, if you find yourself opening/printing the contents of the Slurm log file again and again to keep track of the job’s progress, then instead use tail -f, which will “follow” the file and will print new text as it’s added to the Slurm log file:
# See the last lines of the file, with new contents added in real time
tail -f slurm-fastqc-37615717.outTo exit the tail -f livestream, press Ctrl+C.
Footnotes
Even though technically, one CPU often contains multiple cores.↩︎
Even though technically, one core often contains multiple threads.↩︎
Though there are unfortunately “gaps” between the maximum and minimum amounts of RAM on different nodes…↩︎
For instance, we might be running the FastQC script multiple times, and otherwise those Slurm log files would all have the same name and be overwritten!↩︎
I would avoid having Slurm send you emails upon regular completion for shorter jobs, because you may get inundated with emails and then quickly start ignoring them altogether.↩︎
But keep in mind that batch jobs are sometimes queued/waiting for a while, especially if you submit large amounts of jobs.↩︎