Software management: modules and containers
Week 5 – lecture B

1 Introduction
1.1 Learning goals
In this session about (bioinformatics) software management at OSC and beyond, you will learn:
- How to find and load OSC “modules” to use specific programs
- Why containers and Conda are useful and reproducible alternative ways to use software
- How to find and run Apptainer/Singularity containers to use specific programs
- What the program FastQC does and how you can run it
1.2 Setting up
- At https://ondemand.osc.edu, start a VS Code session in
/fs/ess/PAS2880/users/$USER - Open a terminal and create and then navigate to a
week05dir - Optional: Create a new Markdown file for class notes and save it in your
week05dir
2 Software management (at OSC)
To analyze omics data sets, especially in genomics and transcriptomics, a typical workflow includes running a sequence of specialized bioinformatics software/programs/tools (I will use these terms interchangeably).
For example, let’s take another look at the steps in the RNA-Seq analysis workflow we’ll go through, where each step is associated with a specific program (gray text):
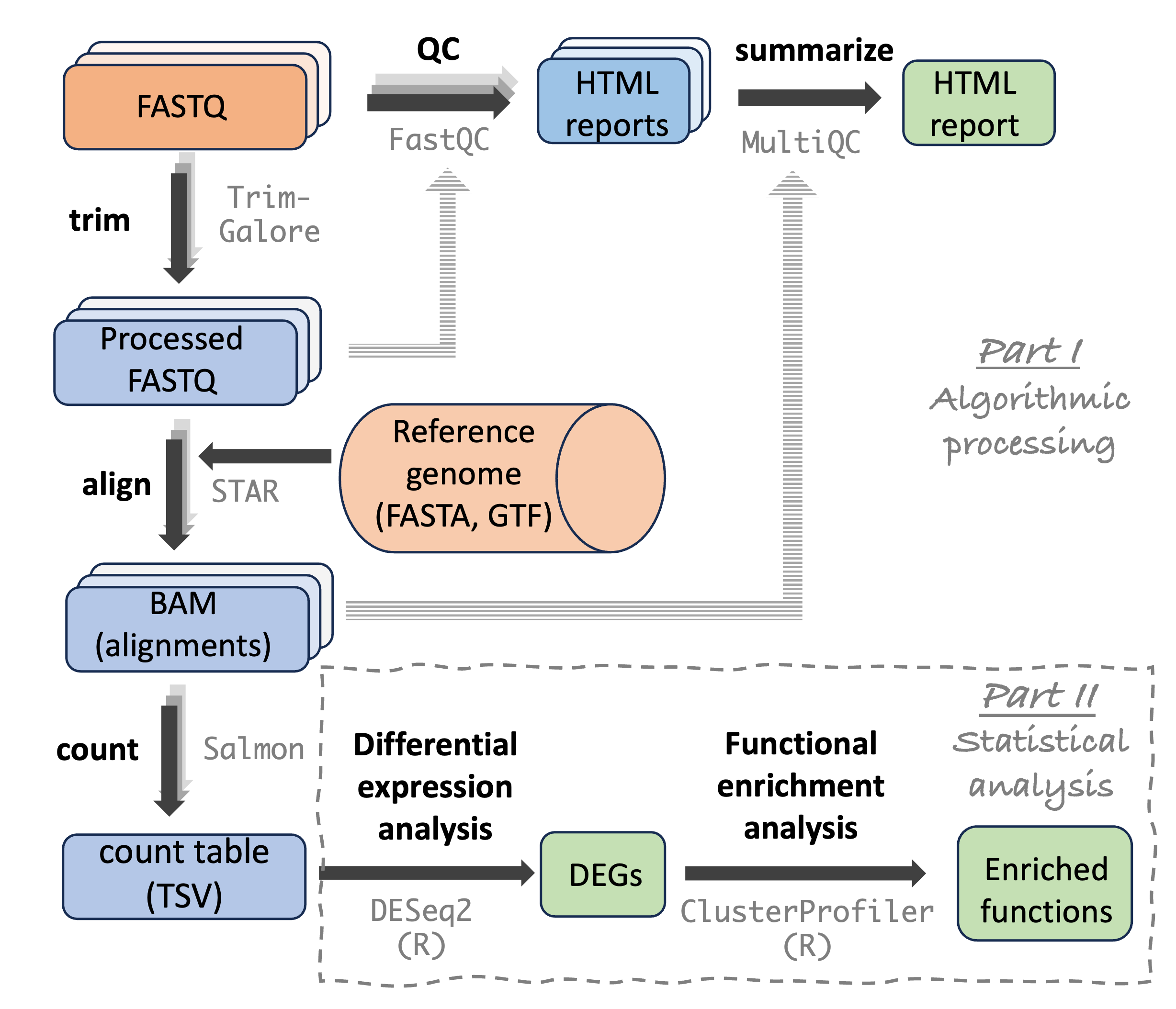
At OSC, there are system-wide installations of a number of such programs. We do need to “load modules” that contain these programs before we can use them. In this session, we will first talk about that process.
However, OSC’s collection of bioinformatics programs is not comprehensive, and programs that are installed may not have the most recent versions available. We therefore need an additional way to get access to bioinformatics programs. But installing software like you are used to on your own computer is not really possible at a supercomputer center like OSC.
The two most best approaches that we can use instead are:
- The Conda software management program, which installs software in “environments” you can load.
- Containers, self-contained software environments that even include large portions of their operating systems.
Conda and containers are useful because:
- At supercomputers like OSC, they bypass issues with “dependencies”1 and “administrator privileges”2 that you may run into when trying to install software in other ways.
- They make it easy to switch between different versions of the same software, or to use mutually incompatible software.
- They are highly reproducible.
In this session, you will learn how to obtain and use containers, while this optional-reading page covers Conda.
Here are two further options to install or get software installed at OSC:
Send an email to OSC Help. They might be able to help you with your installation, or in case of commonly used software, might be willing to perform a system-wide installation (that is, making it available through Lmod /
modulecommands).“Manually” install the software, which in the best case involves downloading a directly functioning binary (executable), but more commonly requires you to “compile” (build) the program. This is sometimes straightforward but can also become extremely tricky, especially at OSC where you don’t have administrator privileges and will often have difficulties with dependencies.
2.1 The FastQC software
As an example piece of bioinformatics software, we will use FastQC (Andrews (2010)) in this session. FastQC is a ubiquitous tool for quality control of FASTQ files: Running FastQC or a similar program is the first step in nearly any high-throughput sequencing project. FastQC is also a good introductory example of a CLI tool.
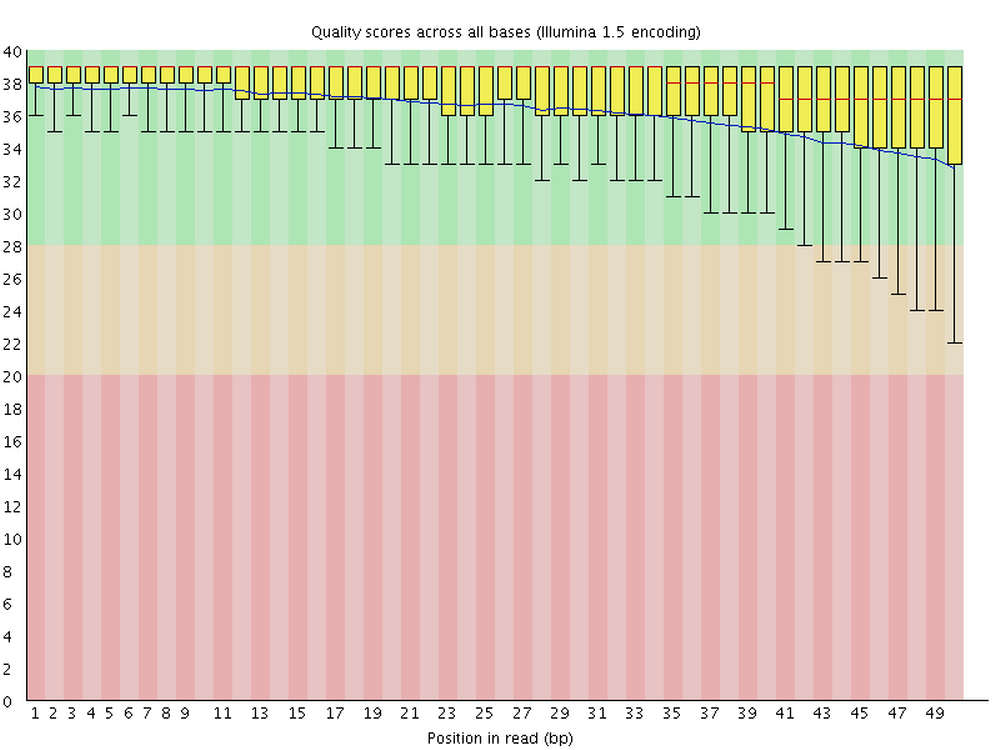
For each FASTQ file that it analyzes, FastQC outputs an HTML file that you can open in your browser with about a dozen graphs showing different QC (Quality Control) metrics. The most important one is this per-base quality score graph:
3 Loading software at OSC with Lmod modules
OSC administrators manage software with the “Lmod” system of software modules. This means that a lot of installed software can only be used after its module is explicitly loaded. That may seem like a drag, but this practice enables the use of different versions of the same software, and of mutually incompatible software, on a single system.
You can load, unload, and search for available software modules using the module command and its sub-commands.
Not all software has to be loaded: besides all the basic Unix commands we have been using, very widely used software like Git, Python, and Apptainer is available to us without having to load anything.
3.1 Searching for available modules
The OSC website has a list of installed software. You can also search for available software in the shell using two subtly different module commands:
module spiderlists all installed modules.module availlists modules that can be directly loaded given the current environment (i.e., when taking into account which other software has been loaded, which can make a difference).
Simply running module spider or module avail spits out complete lists of available programs — it is therefore more useful to add a search term as an argument. For example, try to use module spider to search for fastqc:
module spider fastqc--------------------------------------------------------------------------------
fastqc: fastqc/0.12.1
--------------------------------------------------------------------------------
This module can be loaded directly: module load fastqc/0.12.1
Help:
A quality control tool for high throughput sequence data.We can see that fastqc is available, namely version 0.12.1 (that is, the module is printed as <software>/<version>).
These two search commands are not case-sensitive – but module load, covered below, is case-sensitive
The various OSC clusters mostly but not entirely have the same modules available!
3.2 Loading modules
All other Lmod software functionality is also accessed using module commands. For instance, to make a program available to you, use the load command.
But first, let’s confirm that it is not possible to use FastQC before doing so:
fastqc -vbash: fastqc: command not found...
Failed to search for file: GDBus.Error:org.freedesktop.DBus.Error.NameHasNoOwner: Could not activate remote peer.Notes on the above command:
- The command to run FastQC is
fastqc - Like Unix commands,
fastqchas many options, and we can pass FASTQ files as arguments. - To test for the program’s availability, we will simply run
fastqc -v, which prints the FastQC version.
Now, load the module – specify the module name and its version exactly as module spider printed it:
module load fastqc/0.12.1And then check once again whether you can use FastQC:
fastqc -vFastQC v0.12.1Success! 🥳
3.3 Final notes on modules
Module loading does not persist across shell sessions. Whenever you are in a fresh shell session (including but not limited to after you log into OSC for the day), you’ll have to reload any modules you want to use! And when you run a program that is in a module using a shell script, include the module load command in the script – we’ll see this next week.
Here, we only covered the very basics of using module. To learn a bit more, you can read the boxes below, but in class, we will not go into further detail because we will mostly use containers instead (considering the limitations of these modules and the advantages of containers discussed above).
module commands (Click to expand)
To check which modules are loaded, use
module list. Its output also includes automatically loaded modules — for example, after loadingfastqc/0.12.1, it may show something like the below, with 5 different modules loaded:module listCurrently Loaded Modules: 1) intel-oneapi-mkl/2023.2.0 2) intel/2021.10.0 3) mvapich/3.0 4) modules/sp2024 5) fastqc/0.12.1Occasionally, when you run into conflicting (mutually incompatible) modules, it can be useful to unload modules, which you can do with
module unloadormodule purge:# Unload a specific module with 'module unload': module unload fastqc/0.12.1 # Unload all loaded modules with 'module purge': module purge
module load example: two-step procedure with R (Click to expand)
First, test if R is available without loading anything:
Rbash: R: command not found...Nope. Is it available as a module?
module spider R------------------------------------------------------------------------------------------------------------------------------------------------------------
R: R/4.4.0
------------------------------------------------------------------------------------------------------------------------------------------------------------
Other possible modules matches:
amber, app_code_server, app_jupyter, blender, connectome-workbench, darshan-runtime, darshan-util, deepemhancer, fmriprep, freesurfer, ...
You will need to load all module(s) on any one of the lines below before the "R/4.4.0" module is available to load.
gcc/12.3.0
Help:
R is 'GNU S', a freely available language and environment for
statistical computing and graphics which provides a wide variety of
statistical and graphical techniques: linear and nonlinear modelling,
statistical tests, time series analysis, classification, clustering,
etc. Please consult the R project homepage for further information.
------------------------------------------------------------------------------------------------------------------------------------------------------------
To find other possible module matches execute:
$ module -r spider '.*R.*'Yes, so let’s try to load it:
module load R/4.4.0Lmod has detected the following error: These module(s) or extension(s) exist but cannot be loaded as requested: "R/4.4.0"
You encountered this error for one of the following reasons:
1. Missing version specification: On Ascend, you must specify an available version.
2. Missing required modules: Ensure you have loaded the appropriate compiler and MPI modules.
Try: "module spider R/4.4.0" to view available versions or required modules.
If you need further assistance, please contact oschelp@osc.edu with the subject line "lmod error: R/4.4.0"That didn’t work. Carefully reading the earlier message printed by module spider, it appears you first need to load the module gcc/12.3.0 before you can load R. Specifically, it said:
You will need to load all module(s) on any one of the lines below before the "R/4.4.0" module is available to load.
gcc/12.3.0Therefore, let’s try to load gcc along with R – you can use the command below to load both at the same time:
module load gcc/12.3.0 R/4.4.0Lmod is automatically replacing "intel/2021.10.0" with "gcc/12.3.0".
Due to MODULEPATH changes, the following have been reloaded:
1) mvapich/3.0Finally, let’s check that R works:
RR version 4.4.0 (2024-04-24) -- "Puppy Cup"
Copyright (C) 2024 The R Foundation for Statistical Computing
Platform: x86_64-pc-linux-gnu
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type 'license()' or 'licence()' for distribution details.
Natural language support but running in an English locale
R is a collaborative project with many contributors.
Type 'contributors()' for more information and
'citation()' on how to cite R or R packages in publications.
Type 'demo()' for some demos, 'help()' for on-line help, or
'help.start()' for an HTML browser interface to help.
Type 'q()' to quit R.
> Now you’re in the R console – type q() or use the general keyboard short for exiting Ctrl/Cmd+D to exit R.
Exercise: Modules
Search for the program MultiQC3 – is it available?
Click to see the solution
No, it isn’t available!
module spider multiqcLmod has detected the following error: Unable to find: "multiqc".Search for the program STAR4 – is it available?
Click to see the solution
Yes, it’s available:
module spider star-------------------------------------------------------------------------------- star: star/2.7.10b -------------------------------------------------------------------------------- Other possible modules matches: starccm This module can be loaded directly: module load star/2.7.10b Help: STAR is an ultrafast universal RNA-seq aligner. -------------------------------------------------------------------------------- To find other possible module matches execute: $ module -r spider '.*star.*'Load the STAR module and test whether you can use the program by running
STAR(without adding any options).Click to see the solution
module load star/2.7.10b STARUsage: STAR [options]... --genomeDir /path/to/genome/index/ --readFilesIn R1.fq R2.fq Spliced Transcripts Alignment to a Reference (c) Alexander Dobin, 2009-2022 STAR version=2.7.10b STAR compilation time,server,dir=2025-06-11T15:35:30-04:00 pitzer-rw02.ten.osc.edu:/tmp/kkoepcke/spack/stage/spack-stage-star-2.7.10b-itj64jf32ufsx7x6kbmlpcprqq5xo5 in/spack-src/source For more details see: <https://github.com/alexdobin/STAR> <https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf> To list all parameters, run STAR --helpBonus: Open a Unix shell on the Cardinal cluster: in OnDemand, click “Clusters” in the blue top bar, and then “Cardinal Shell Access”. Also check there whether STAR is available and which versions – does it differ from the situation in Pitzer?
Click for the solution
Yes, two different versions are available on Cardinal, including a more recent version than on Pitzer:
module spider star----------------------------------------------------------------------------- star: -------------------------------------------------------------------------------- Versions: star/2.7.10b star/2.7.11b Other possible modules matches: starccm
4 Using Apptainer containers
4.1 Introduction
As mentioned above, because many programs needed for omics data analysis are not available through OSC modules, you need an alternative way to access these.
Conda and containers are both great options: here, we will go over containers, while optional at-home reading covers Conda. These two approaches are quite different both conceptually and operationally. Conda is similar to OSC’s modules, except that you can also install things yourself. Containers, on the other hand, are not installed or loaded5: typical usage involves simply downloading a file and using it.
Key advantages of Conda are that:
- Once software is installed, using it is just like using other software that has been installed. With containers, on the other hand, you essentially need a long “prefix” to your commands.
- It is a bit easier to create custom environments with multiple programs or even with programs you installed “manually” (not with Conda commands)
Key advantages of containers are that:
- They are more portable, i.e. likely to work across different computers. For that and a few other reasons, they’re overall a tad more reproducible than Conda.
- It is quicker to download a container than to install software with Conda, and Conda installation sometimes fails altogether
- Conda environments often consists of tens of thousands of files, and you may well hit the file number quota (yes, these exist at OSC!) when you have a number of Conda environments.
- At OSC, Singularity doesn’t require the loading of a module unlike Conda
- With the container usage you’ll learn, you need to remember fewer commands than with Conda
One thing that currently does not differ is:
- The same set of programs is available through Conda and as containers – basically, their repositories are tightly linked
Among container platforms, Apptainer (formerly, and in some cases still, known as Singularity) and especially Docker are most widely used. At supercomputers like OSC, however, only Apptainer containers can be used6. Some Apptainer container terminology:
- Container image: a binary file that contains the container application (
.sifextension) - Container (sensu stricto): a running container image
- Definition file: a plain text file that contains the recipe to build a container image
4.2 How we’ll use containers
Here, you will learn how to find container images online and use them at OSC. We will not cover writing Definition files to create your own custom containers, because this is very rarely needed given recent developments in online container availability. The website we’ll use has, or can be asked to create, container images for nearly all open-source bioinformatics software!
As such, for us, the first step to using a container is simply finding/obtaining one. The second step is to run an appropriate command to access the software installed inside the container. We’ll go over these steps one-by-one.
4.3 Obtaining a container image
Several online repositories with publicly available container images exist, but I strongly recommend Seqera Containers.
Seqera Containers
Seqera Containers is currently the most reliable website to find and download available containers. It is actually more than just a repository: when a container with the program(s) you request is not available in the repository, it will build the container for you. It allows you to select:
- Any recent version of software that’s available – and nearly all open-source bioinformatics software is
- Any combination of software that’s not mutually incompatible
Then, it will see if a container with your exact requirements is already available and if not, it will build it and make it available.
The following applies to both containers and Conda:
- Only free, open-source software is available
- It is a good idea to avoid proprietary/paid software anyway!
- Rarely, academic free software has restrictive licensing that prevents it from being available
- You may run into an old or “low-key” program/script that is not available. I would take this as a sign that it may be better to avoid that program anyway – in practice, it is likely to be outdated, poorly maintained, and/or poorly documented.
Finding a container, step-by-step
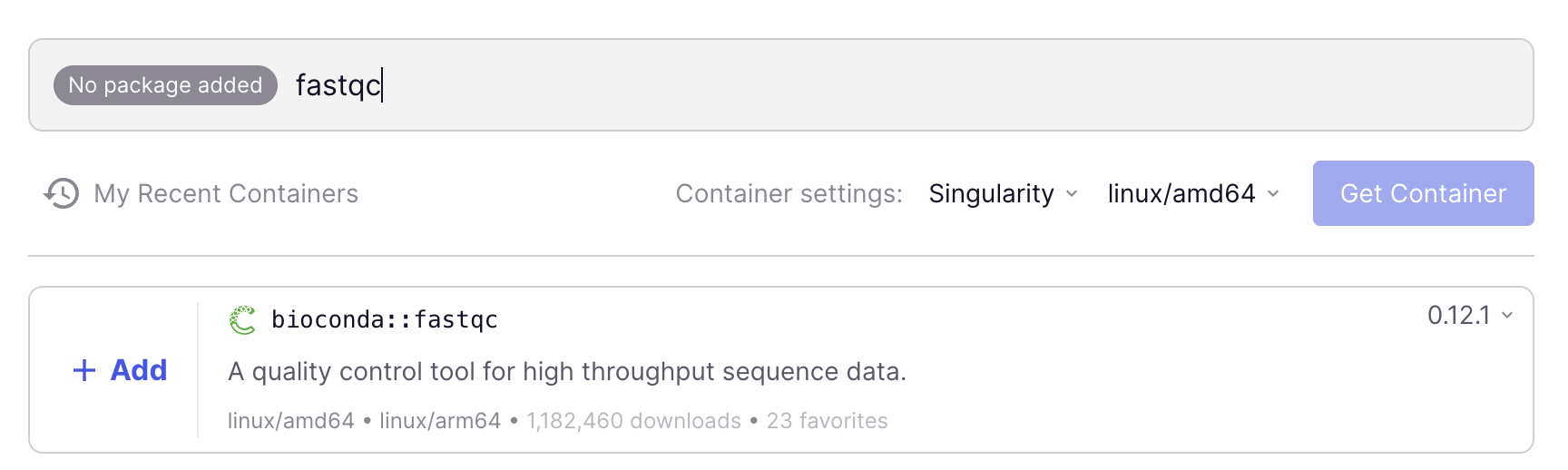
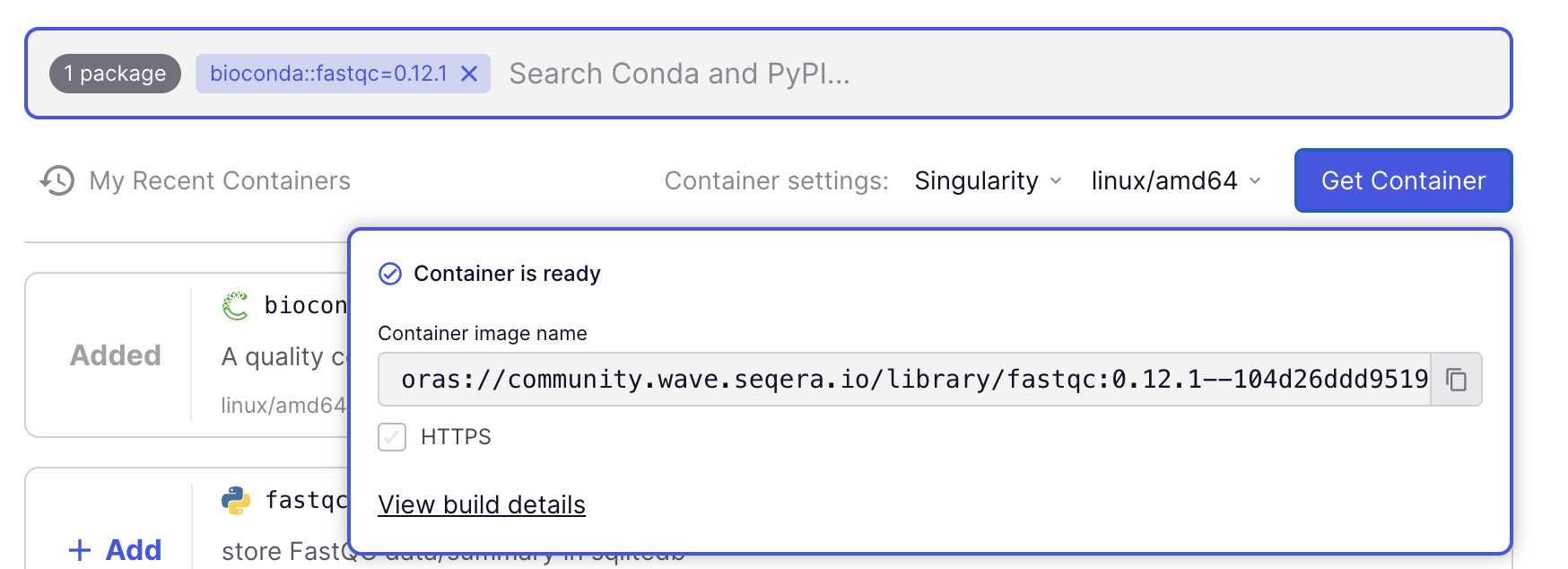
Here is a step-by-step procedure to find a container image – again using FastQC version 0.12.1 as the example, but the procedure would be the same for any other available program:
In the search box below “Containers”, type
fastqc.After waiting for the list to populate, the top hit should be
bioconda::fastqc, with version0.12.1selected (the most recent version is always selected by default)
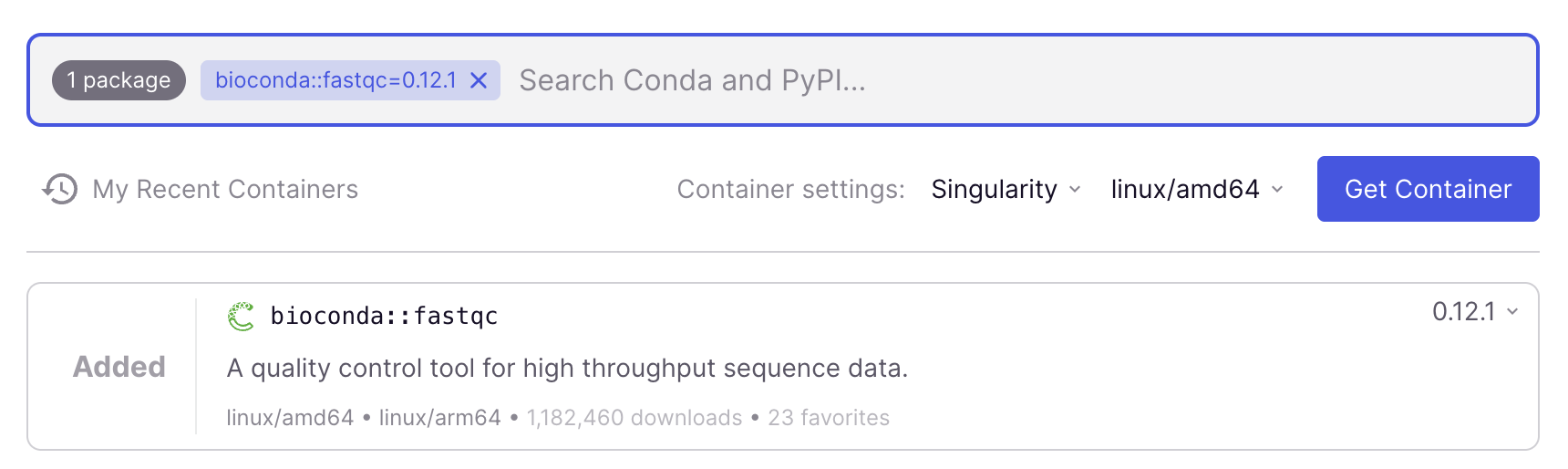
Add FastQC to your container request by clicking the blue
+ Addbutton. At this point, you could add additional programs, but for this example, we are happy with a container that only has FastQC.In “Container settings” below the search box, change from
DockertoSingularity(and leave the other dropdown as is –linux/amd64should be selected).
Get a link to the container image by clicking the blue
Get Containerbutton.Copy the link (URL/URI) to the container image that should appear immediately (use the copy icon on the far left). The link should be:
oras://community.wave.seqera.io/library/fastqc:0.12.1--104d26ddd9519960
A few more tips on selecting programs for your container on this website:
- If we had wanted a different (older) version of FastQC, we could have used the dropdown menu on the right:
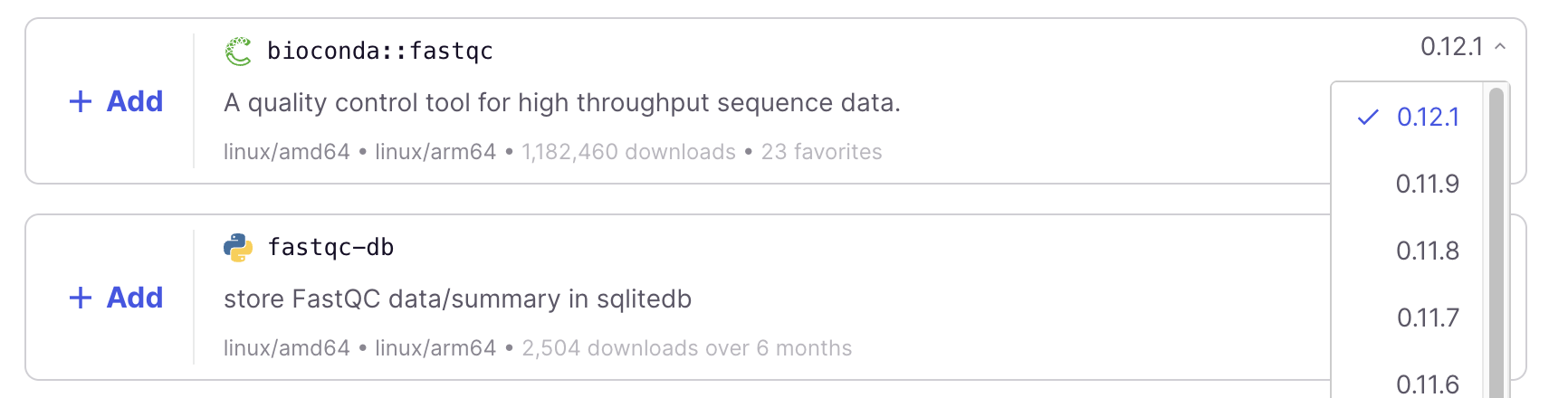
- You will always want to pick Conda-based search hits, which you can recognize in two ways:
- The program name will be prefixed by
bioconda::(most commonly) orconda-forge::. - Its logo, the green open circle (snake) shown in the screenshot above
- The program name will be prefixed by
In this case, after we clicked Get Container, the pop-up box should have immediately said “Container is ready”. That means the container image was already available in the repository.
In other cases, however, the container may still need to be built, and the box should say “Fetching container” for a while. This could take one or a few minutes, and even though the URL is made available immediately and won’t change, you can only download/use the container once the “Container is ready”. If you use an URL before the container is ready, you get this error:
apptainer exec oras://community.wave.seqera.io/library/multiqc_trim-galore:15a1e9f26daf266f \
multiqc -vFATAL: Unable to handle oras://community.wave.seqera.io/library/multiqc_trim-galore:15a1e9f26daf266f uri: failed to get checksum for oras://community.wave.seqera.io/library/multiqc_trim-galore:15a1e9f26daf266f: GET https://community.wave.seqera.io/v2/library/multiqc_trim-galore/manifests/15a1e9f26daf266f: MANIFEST_UNKNOWN: manifest unknown; map[Tag:15a1e9f26daf266f]4.4 Running a container image
To run a command in a container, the general syntax is:
apptainer exec <container-URL> <command>For example, if the command (<command> above) you want to run is fastqc -v:
apptainer exec <container-URL> fastqc -vSo, you need to preface your “regular” command with apptainer exec <container-URL>. Now with the actual link to the container:
apptainer exec oras://community.wave.seqera.io/library/fastqc:0.12.1--104d26ddd9519960 \
fastqc -vINFO: Downloading oras image
384.0MiB / 384.0MiB [======================================================================================================================] 100 % 51.8 MiB/s 0s
INFO: gocryptfs not found, will not be able to use gocryptfs
FastQC v0.12.1\
Above, I broke the command up across two lines, which can be done by ending the first line with a \. With a \, the shell understands that the command is still incomplete despite the newline. Using \ to break up your commands across multiple lines helps with readability for long commands like this one.
The first time you run this command, the container image will be downloaded to a default location (see box below), as you can see in he output above — this will take a minute or so.
4.5 The container image cache (storage)
The easiest way to run a container image you already downloaded is to simply keep re-use the exact same command with the URL to the container (!): Apptainer will realize that the image has already been downloaded and will automatically use the downloaded version:
apptainer exec oras://community.wave.seqera.io/library/fastqc:0.12.1--104d26ddd9519960 \
fastqc -vINFO: Using cached SIF image
INFO: gocryptfs not found, will not be able to use gocryptfs
FastQC v0.12.1- The
Using cached SIF imageline tells you that it is using a previously-downloaded SIF file - You will keep seeing the
gocryptfs not foundmessage, but this is nothing to worry about.
By default, containers are downloaded to a hidden directory in your Home dir, ~/.apptainer/cache. That is generally fine, but you can change it for your current shell session by setting the $APPTAINER_CACHEDIR environment variable, e.g.:
export APPTAINER_CACHEDIR=/fs/scratch/PAS2880/$USER/containers(To permamently change this location, you would need to add this line to your ~/.bashrc shell configuration file. We won’t cover this file in class.)
You can check what’s in your cache as follows – though this is only informative in terms of how many containers are there and how much space they take up, because the container names are very cryptic:
apptainer cache list -vNAME DATE CREATED SIZE TYPE
sha256:e0c976cb2eca5fe 2025-09-20 19:08:51 383.99 MiB oras
There are 1 container file(s) using 383.99 MiB and 0 oci blob file(s) using 0.00 KiB of space
Total space used: 383.99 MiB5 A practical example: running FastQC on a FASTQ file
To analyze a FASTQ file with default FastQC settings, the command is simply fastqc followed by the FASTQ file path – and we could use one of the FASTQ files in our Garrigos practice data set:
# [Don't run this]
apptainer exec oras://community.wave.seqera.io/library/fastqc:0.12.1--104d26ddd9519960 \
fastqc ../garrigos-data/fastq/ERR10802863_R1.fastq.gzTo shorten subsequent commands, let’s start by assigning the container link and FASTQ file path to variables:
FASTQC_APPT=oras://community.wave.seqera.io/library/fastqc:0.12.1--104d26ddd9519960
FASTQ_R1=../garrigos-data/fastq/ERR10802863_R1.fastq.gzecho "$FASTQC_APPT"
ls -lh "$FASTQ_R1"oras://community.wave.seqera.io/library/fastqc:0.12.1--104d26ddd9519960
-rw-rw----+ 1 jelmer PAS0471 21M Sep 9 13:46 ../garrigos-data/fastq/ERR10802863_R1.fastq.gzWith that, the above command becomes:
# [Don't run this]
apptainer exec "$FASTQC_APPT" \
fastqc "$FASTQ_R1"However, an annoying default behavior of FastQC is that it writes its output files in the same dir that contains the input FASTQ files — this means mixing your raw data with your results, which we don’t want!
5.1 Changing the output directory?
To figure out how to change that behavior, consider that many standard Unix commands and bioinformatics tools alike have an option -h and/or --help to print usage information to the screen. Let’s try that:
apptainer exec "$FASTQC_APPT" \
fastqc -hINFO: Using cached SIF image
INFO: gocryptfs not found, will not be able to use gocryptfs
FastQC - A high throughput sequence QC analysis tool
SYNOPSIS
fastqc seqfile1 seqfile2 .. seqfileN
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam]
[-c contaminant file] seqfile1 .. seqfileN
# [...output truncated...]Exercise: FastQC help and output dir
Look at FastQC’s help info, and figure out which option can be used to specify a custom output directory.
Click for the solution
The option to do this is -o/--outdir:
fastqc -h -o --outdir Create all output files in the specified output directory.
Please note that this directory must exist as the program
will not create it. If this option is not set then the
output file for each sequence file is created in the same
directory as the sequence file which was processed.5.2 The final FastQC command
With the added --outdir (or -o) option, try to run the following FastQC command:
# We'll have to first create the outdir ourselves, in this case:
mkdir -p results/fastqc
# Now we run FastQC:
apptainer exec "$FASTQC_APPT" \
fastqc --outdir results/fastqc "$FASTQ_R1"Started analysis of ERR10802863_R1.fastq.gz
Approx 5% complete for ERR10802863_R1.fastq.gz
Approx 10% complete for ERR10802863_R1.fastq.gz
Approx 15% complete for ERR10802863_R1.fastq.gz
[...truncated...]
Analysis complete for ERR10802863_R1.fastq.gzSuccess! Above, FastQC printed some “logging” information to screen, telling us about its progress. The main output, however, is in the output dir we specified:
- A
.zipfile, which contains tables with FastQC’s data summaries - An
.html(HTML) file, which contains the graphs we’d like to see
ls -lh results/fastqctotal 512K
-rw-rw----+ 1 jelmer PAS0471 241K Mar 21 09:53 ERR10802863_R1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 256K Mar 21 09:53 ERR10802863_R1_fastqc.zipFastQC allows us to specify the output directory, but not the output file names: these will be automatically determined based on the input file name(s). This kind of behavior is fairly common for bioinformatics programs, since they will often produce multiple output files.
Exercise: Another FastQC run
Run FastQC for the corresponding R2 FASTQ file. Would you use the same output dir or a different one?
Click for the solution
It makes sense to use the same output dir. This is because, as you could see above, the output file names have the input file identifiers in them. As such, because we don’t need to worry about overwriting files, it will be more convenient to have all results in a single dir.
To run FastQC for the R2 (reverse-read) file:
FASTQ_R2=../garrigos-data/fastq/ERR10802863_R2.fastq.gz
apptainer exec "$FASTQC_APPT" \
fastqc --outdir results/fastqc "$FASTQ_R2"Started analysis of ERR10802863_R2.fastq.gz
Approx 5% complete for ERR10802863_R2.fastq.gz
Approx 10% complete for ERR10802863_R2.fastq.gz
Approx 15% complete for ERR10802863_R2.fastq.gz
[...truncated...]
Analysis complete for ERR10802863_R2.fastq.gzls -lh results/fastqctotal 1008K
-rw-rw----+ 1 jelmer PAS0471 241K Mar 21 09:53 ERR10802863_R1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 256K Mar 21 09:53 ERR10802863_R1_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 234K Mar 21 09:55 ERR10802863_R2_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 244K Mar 21 09:55 ERR10802863_R2_fastqc.zipNow, we have four files: two for each of our preceding successful FastQC runs.
Exercise: Download the HTML files and open them
Unfortunately, it’s tricky to view HTML files in VS Code – or at least it is in the current Code Server version at OSC.
Therefore, to look at the FastQC HTML files, you should first download them to your computer.
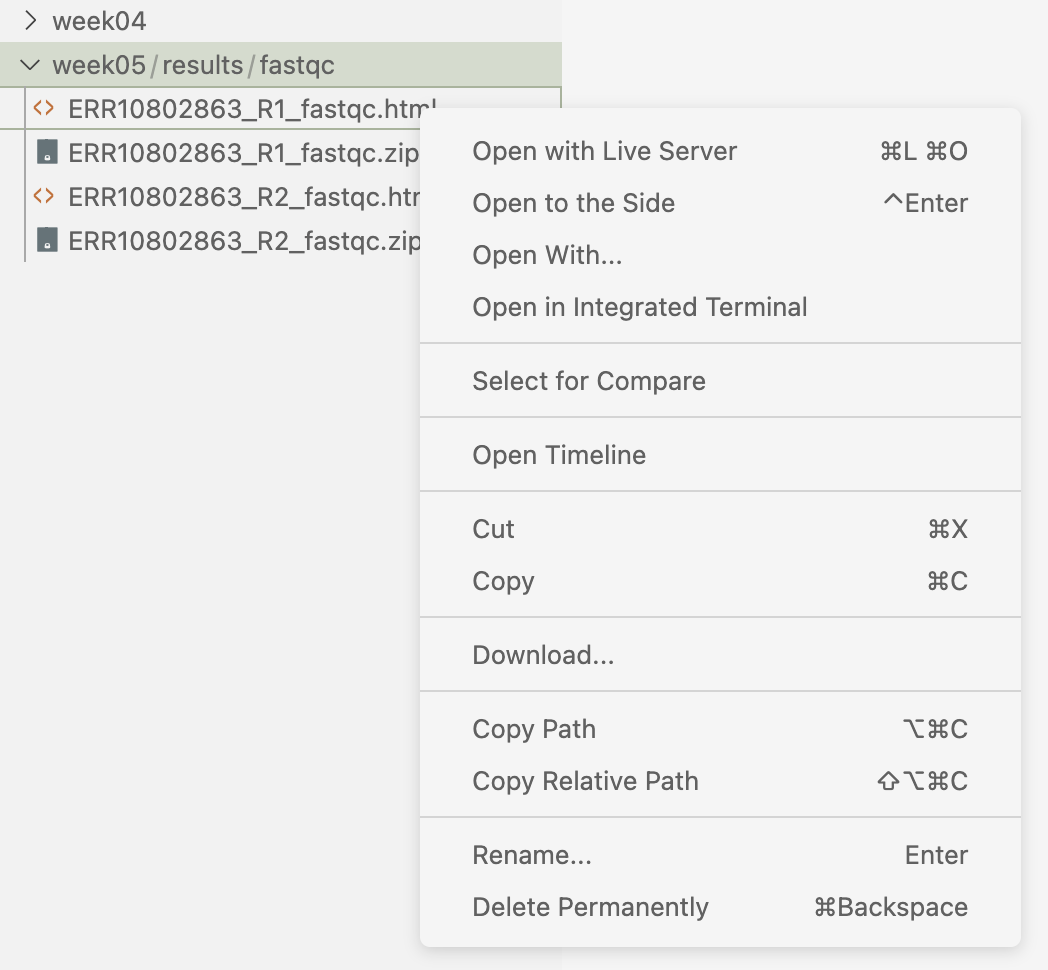
We’ll cover more scalable methods for downloading files from and uploading them to OSC in a couple of weeks, but here is a quick method when you just need one or few files: in the VS Code file explorer in the side bar, you can download a file by right-clicking on it and then selecting the “Download…” option.
Your turn:
Download the two FastQC HTML files to your computer.
In your computer’s file browser, navigate to the folder you downloaded the files to, and click on each of them. This should open the files in your default web browser.
Explore the files. Which graphs do you and which do you not understand?
Are there noticeable differences between the two files (i.e. between the forward and reverse reads for the same sample)?
6 In closing
In research with full-size datasets, you typically don’t want to run bioinformatics programs like FastQC “interactively” like we just did, i.e. by executing the command directly in the terminal.
Instead, it is preferable to write small shell scripts to run such programs, and then submit those scripts as so-called “batch jobs” at OSC. In the next two weeks, you will learn why that is the case and how you can do this:
- Next week, you’ll learn how to write shell scripts and loop over files
- In two weeks, you’ll learn how submit shell scripts as batch jobs.
References
Footnotes
Other software upon which the software that you are trying to install depends.↩︎
When your personal computer asks you to “authenticate” while you are installing something, you are authenticating yourself as a user with administrator privileges. At OSC, you don’t have such privileges.↩︎
MultiQC summarizes the outputs of FastQC and many other bioinformatics programs.↩︎
STAR is an RNA-Seq aligner that we will use to align the reads from our example dataset to our reference genome.↩︎
Though it is possible to create containers yourself and install software into them – but we will not cover that.↩︎
But luckily, the Apptainer program can work with Docker container images: it will convert them on the fly.↩︎